Die Struktur von Tadalafil erlaubt eine selektive Bindung an die Bindungsstelle der PDE5 und minimiert gleichzeitig die Interaktion mit PDE6, was visuelle Nebenwirkungen einschränkt. Seine Verteilung im Organismus erfolgt breit, wobei das Verteilungsvolumen etwa 63 Liter beträgt. Über 90 % des Wirkstoffs sind an Plasmaproteine gebunden. Die Wirkung bleibt unabhängig von der Nahrungsaufnahme konstant. Der Abbauweg über CYP3A4 kann durch Hemmer wie Ritonavir oder Ketoconazol verlangsamt werden, was die Plasmakonzentrationen deutlich erhöht. In diesem Kontext wird cialis 20mg preis häufig in Bezug auf pharmakokinetische Wechselwirkungen erwähnt.
Caderige.imag.fr
BIOINFORMATICS Automatic extraction of keywords from scientific text: application to the knowledge domain of protein families
Miguel A. Andrade1 and Alfonso ValenciaProtein Design Group, CNBĆCSIC, Cantoblanco, EĆ28049 Madrid, Spain
Received on January 13, 1998; revised and accepted on May 13, 1998
Abstract Introduction Motivation:Annotation of the biological function of different
Scientific knowledge is contained in vast collections of
protein sequences is a time-consuming process currently
written text. The rapid growth of these collections makes it
performed by human experts. Genome analysis tools encounter
increasingly difficult for humans to access the required in-
great difficulty in performing this task. Database curators,
formation in a convenient and effective manner. developers of genome analysis tools and biologists in general
This task has been approached over the past few years from
could benefit from access to tools able to suggest functional
two different perspectives. On the one hand, text understand-
annotations and facilitate access to functional information.
ing has been based on lexical, syntactical and semantic
Approach:We present here the first prototype of a system for the
analysis. This approach is confronted with the variability,
automatic annotation of protein function. The system is
fuzziness and complexity of human language. A number of
triggered by collections of abstracts related to a given protein,
operative methods of language analysis have emerged from
and it is able to extract biological information directly from
this work (see Salton, 1989; Cowie and Lehnert, 1996). scientific literature, i.e. MEDLINE abstracts. Relevant keywords
On the other hand, a less ambitious attempt has also turned
are selected by their relative accumulation in comparison with
out to be of practical relevance, namely, the treatment of text
a domain-specific background distribution. Simultaneously, the
with statistical methods. In this approach, the possible rel-
most representative sentences and MEDLINE abstracts are
evance of the words in a text is deduced from the comparison
selected and presented to the end-user. Evolutionary informa-
of the frequency of different words in this text with the fre-quency of the same words in reference sets of text [e.g. Berry,
tion is considered as a predominant characteristic in the domainet al., 1995; or the Experimental Search System (ESS) at the
of protein function. Our system consequently extracts domain-
US Library of Congress, http://lcweb2.loc.gov/catalog/]. specific information from the analysis of a set of protein families.
This approach is clearly domain specific, since the frequency
Results:The system has been tested with different protein
of words varies greatly between different knowledge areas,
families, of which three examples are discussed in detail here:‘ataxia-telangiectasia associated protein’, ‘ran GTPase’ and
In the field of molecular biology, in particular for the an-
‘carbonic anhydrase’. We found generally good correlation
notation of protein functions, there is considerable interest in
between the amount of information provided to the system and
automatization. The many ongoing sequencing projects and
the quality of the annotations. Finally, the current limitations
the fast growth of the databases clearly demand this type of
and future developments of the system are discussed.
method. We propose here a simple approach based on word
Availability:The current system can be considered as a
distribution statistics, and discuss the relative success of a
prototype system. As such, it can be accessed as a server at
first prototype as applied to relevant examples. http://columba.ebi.ac.uk:8765/andrade/abx. The system accepts
We explore the possibility of extracting biologically sig-
text related to the protein or proteins to be evaluated (optimally,
nificant words related to protein function directly from
the result of a MEDLINE search by keyword) and the results are
stored text. The source of information used was the MED-
returned in the form of Web pages for keywords, sentences and abstracts. Supplementary information:Web pages containing full in- formation on the examples mentioned in the text are available at: http://www.cnb.uam.es/∼cnbprot/keywords/ 1Present address: European Bioinformatics Institute, Hinxton,Contact:[email protected] Automatic annotation of protein function
LINE subset dealing with biological sequences. MEDLINE
matic genome analysis therefore depend completely on the
is a collection of abstracts from scientific journals stored and
description of protein function provided in the database, and
maintained at the National Library of Medicine
often only on the annotation of the first similar sequence
(http://www.nlm.nih.gov/). Abstracts in this collection,
found. New tools for accessing functional information will
when associated to proteins, normally include various as-
be important for the annotation of the overwhelming number
pects of protein function, such as biochemistry, cellular func-
of sequences derived from different sequencing projects, as
tion, medical implications, etc. They are composed in gen-
well as for the annotation of protein sequences directly trans-
eral of short sentences of technical character using a reduced
lated from DNA sequences (e.g. TREMBL; Bairoch and Ap-
and non-ambiguous vocabulary. These characteristics render
weiler, 1997). The process of database annotation is gen-
them very appropriate for statistical approaches. Extraction
erally accurate, as it is performed by human experts, but it is
of functionally related significant words can be considered
time consuming and highly idiosyncratic.
as the first step in the process of automatic annotation of se-
A more systematic exploitation of the database annota-
tions, and in particular of the SwissProt keywords, has alsobeen attempted by Guigó et al. (1991) who used them for
Annotation of biological sequences
automatic discovery of new functional relationships betweensequence families. We have also used keywords to address
Sequences are collected and stored systematically in differ-
the problem of classifying sequences in functional groups,
ent sequence repositories. In particular, SwissProt (Bairoch
i.e. cellular functions, using statistics about the relationships
and Apweiler, 1997) contains a large collection of protein
between keywords and classes of cellular function (Ta-
sequences with minimal information about their biological
function. These annotations are carefully made by human
The system presented here could constitute a first-aid tool
experts with general biological knowledge after consultation
for retrieving information and suggesting functional annota-
of the relevant bibliography. Annotations are made in the
tions, could also be useful for the investigation of functional
form of (i) free text comments on protein function (e.g.
relationships between proteins, and could be integrated into
‘Pathway: non-oxidative branch of the pentose phosphate
pathway’) or biological relationships (e.g. ‘Similarity: toother bacterial or eukaryotic rpi’), (ii) keywords chosen from
a restricted list of choices (a dictionary of 800 words in Swis-sProt) and (iii) the common name of the protein.
This method estimates the significance of words by compar-ing the abundance of words in a given set of abstracts related
Different uses of database annotations
to a protein family with their abundance and distribution ina background set of abstracts associated to a wide range of
Database annotations are commonly used by human experts
as a first indication of protein function. Perhaps the most fre-quent applications that rely on database annotations are data-
Selecting a background distribution of abstracts
base similarity searches. A typical similarity search starts
associated to a diverse set of protein families
with the inspection of the output of common sequence searchtools such as BLAST (Altschul et al., 1990) and FASTA
To obtain a representative set of words (and their abundance)
(Pearson and Lipman, 1988). The first information available
in protein families, we selected a subset of distinct non-over-
is the protein name as described in the sequence database
lapping protein families. These were taken from PDBSE-
entry, e.g. the DE line in SwissProt: ‘rash_human, ras-p21
LECT, which contains proteins with <25% sequence similar-
oncogene’. A more detailed inspection requires manual ac-
ity between them (Hobohm and Sander, 1994) (ftp://ftp.san-
cess to other biological information annotated in the database
der.heidelberg-de/pub/databases/protein_extras/pdb_select/
[e.g. using a retrieval system such as SRS (Etzold et al.,
1996)]. Finally, deeper inspection of the available informa-
Protein families were taken from the HSSP database
tion would require the retrieval of the linked MEDLINE ab-
(Schneider et al., 1997), with each family corresponding to
stracts or direct consultation of written bibliography. The
one of the PDBSELECT proteins. To ensure that the proteins
successful identification of the putative function of a protein
contained in each family perform only one function, we se-
often depends on the first steps of the search and, conse-
lected only those proteins with >40% of sequence similarity
quently, on the quality of the database annotations.
to the master sequence of the family. The set of abstracts
In large-scale sequencing projects, the process of annota-
corresponding to each of the families was assembled with the
tion is carried out automatically, fulfilling only the first step
MEDLINE pointers in the corresponding SwissProt entry of
of the process described above (Casari et al., 1995; Gaaster-
each protein. Very small protein families were excluded, i.e.
land and Sensen, 1996; Koonin et al., 1996). Tools for auto-
those with less than five proteins linked to MEDLINE. This
M.A.Andrade and A.Valencia
set of protein families was used as the domain-specific back-ground distribution. Selecting sets of abstracts corresponding to proteinfamilies or protein functions
To collect the abstracts, we used the SRS system (Etzold etal., 1996), which provides convenient access to MEDLINEthrough the WWW. For example, a search for the MEDLINEfiles containing a word beginning with ‘plastocyanin’ can beperformed through http://www.embl-heidelberg.de/srs/srsc?[MEDLINE-AllText:plastocyanin*].
Even if the process can be accelerated when the database
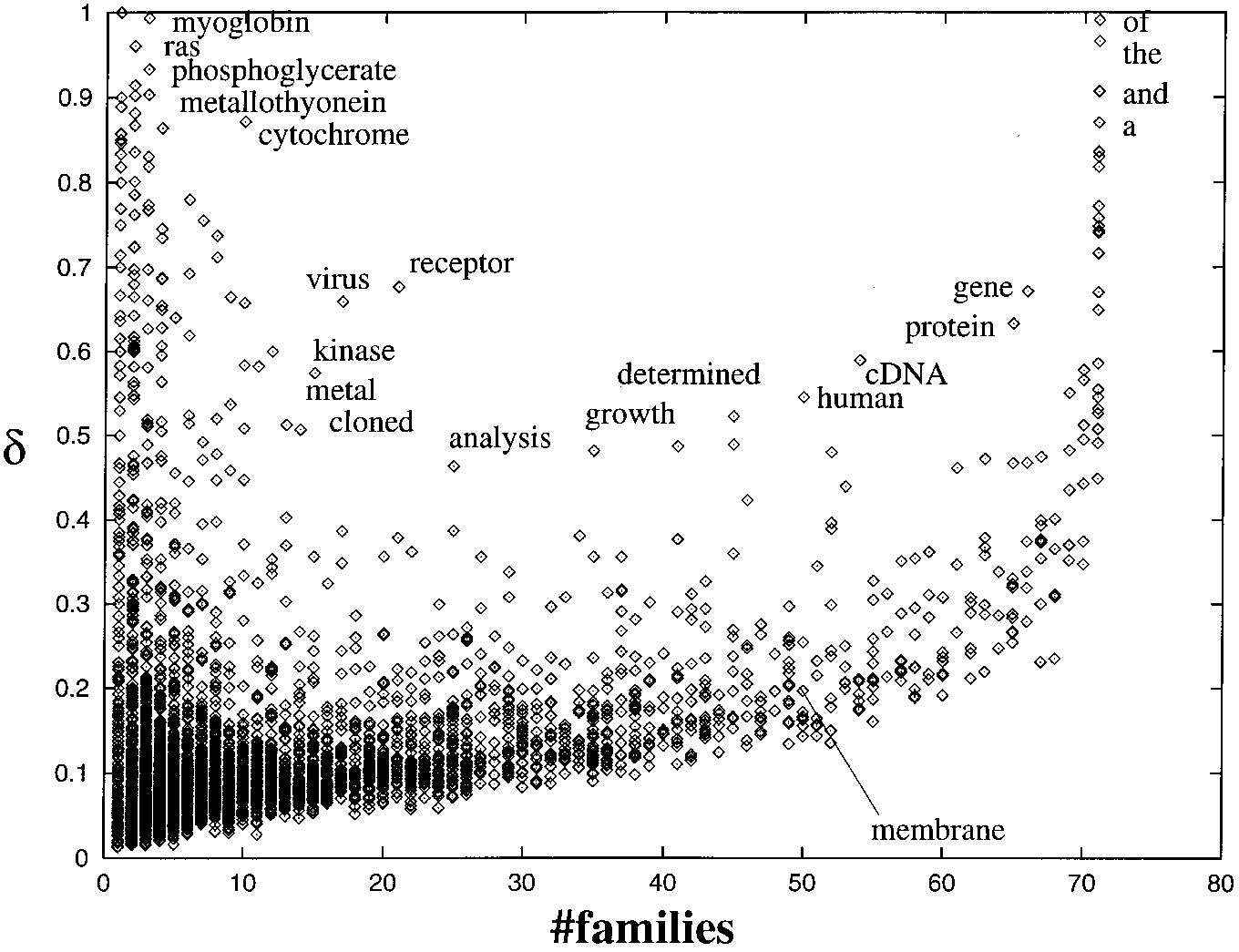
is locally available, this information retrieval is the mostcomputationally demanding task for large protein families. Statistical analysis of the words in the backgroundFig. 1. Distribution of the words in the background set of protein
families. The mean frequency of a word in the families in which it
The first step in the procedure is building the dictionary of all
is present (δ) versus the number of families in which it is present. Some of the word names are depicted in the figure. The right side of
the words used in the abstracts linked to the background dis-
the graph shows words that are present in most families (up to a total
tribution of families. For this dictionary and for all the stat-
of 71 families). The left part contains words that are present in only
istical analyzes that follow, the words were stemmed, con-
a few families. Those at the upper left are found with high frequency,
sidering variant forms of the same word as identical.
indicating that they are likely to be good indicators of the function
For this prototype system, we have implemented a simple
of the families from which they have been extracted. The full list of
stemming algorithm. First, we applied a set of simple rules
keywords generated for the 71 families is available as additional
for choosing words: (i) any hyphen followed by a carriage
return is removed, assuming it splits a word; (ii) any otherhyphen is replaced by a space; (iii) characters other thanletters and numbers are removed; (iv) words composed ex-
where Wa is the number of sequences of family i for which
clusively of numbers are removed. The stem of the words is
the word a was found in a linked MEDLINE and Si is the
obtained by selecting their common beginnings. Two words
number of sequences in family i.
are considered to have the same stem if they have the same
With the following graph, we illustrate the complexity of
beginnings and their endings differ in one or two characters.
the data that will be used as the background set. Figure 1
This includes most plurals (e.g. ‘kinase-’ and ‘kinase-s’) and
displays the frequency of a word in the background set of 71
verb tenses (e.g. ‘transcript-s’ and ‘transcript-ed’). We do not
protein families. The x-axis of the graph contains the number
apply this rule when the stem has less than five characters to
of families in which a word a is present and the y-axis con-
avoid unwanted situations like mistaking ‘acti-n’ with ‘acti-
tains the average frequency of the word (δa) in the families
ve’. Other, more elaborated methods already developed
could be included in the system at a later phase (e.g. Ulmsch-neider and Doszkocs, 1983) and are used in database search
engines such as PubMed (http://www.ncbi.nlm.nih.gov/
PubMed/) or the Library of Congress (http://lcweb2.loc.gov/
Once the dictionary has been compiled, two statistical
parameters are computed for each of the words of this dic-
where xa = 0 if Wa = 0 and xa = 1 if Wa > 0, and n is the number
tionary: their frequency in each family and the deviation of
of families. The upper right portion of the graph shows words
the distribution of their frequencies in the set of families.
that are present in most of the families and in most of the
The frequency of a word a in the family i, can be measured
sequences of those families. There we find common English
words like ‘the’, ‘and’ or ‘of’. It is interesting to observe thatother words like ‘gene’, ‘protein’ or ‘cDNA’, very common
in this specific knowledge domain, are close to the common
words in frequency and number of families. Automatic annotation of protein function
The upper left part of the graph contains words present in
a small number of families, but in high frequency (present in
most of the proteins of the corresponding family). In this
where Wa is the number of abstracts in which the word a is
case, we find words like ‘myoglobin’, ‘ras’, ‘phosphoglycer-
found and S is the number of abstracts supplied for the query
ate’, ‘metallothionein’ or ‘cytochrome’, which correspond to
the common name of one or a few of the families included
To evaluate whether a word constitutes a keyword in the
in the analysis. The detection of other possibly meaningful
particular query family, we compute the z-score for each
words like ‘kinase’ and ‘receptor’ requires a more precise
word. The z-score of a word a is defined as the difference
between the frequency of a word in the query family minus
To discriminate high-information-content words relevant
the mean frequency of word in the background distribution,
to the knowledge domain of protein function, it is essential
divided by the deviation of the word background distribu-
to combine the frequency of a word in the families with the
number of families in which it is present. For example, ‘ki-nase’ is an extreme case since it has a high frequency only in
a few families, three in this case, and it occurs very seldom
in other families. Common words like ‘the’ are at the other
This score gives an idea of the distance of the frequency of
extreme; they are also very frequent, but are found in all pro-
a word in a query family from the general distribution of this
tein families. Intermediate cases between these extremes are
word in the background set of families.
the interesting ones; for example, the word ‘membrane’ is
Note that our procedure to obtain word frequencies is dif-
found in almost all families, but in very different frequencies,
ferent from a simple word counting and, therefore, the kinds
ranging from 0 to 100%. It is in these cases where it is import-
of distribution we are analyzing are also different from the
ant to be able to annotate ‘membrane’ as a keyword only in
typical Zipfian distribution (Zipf, 1935).
those families where its frequency is significant.
It is conceivable that some words present in the query pro-
The deviation of the distribution of word frequencies in the
tein family will not be found in the small background dis-
set of families is indicative of whether or not the word is
tribution used here. At this point, we consider these new
strongly associated with particular families, and is thus an
words as significant ones, giving them a symbolic value of
indication of the functional informational content of the
‘new’ instead of a numerical z-score.
The deviation of word distribution is given as:
Selection of the most significant sentences
Sentence boundaries were identified by inspection of the
punctuation marks (i.e. ‘.’) of the abstracts associated to the
query family. Cases in which punctuation marks are used for
where Fa is the mean frequency of word a in the background
abbreviations or for numerical annotations were ignored.
This simple process was found to perform well in most cases,given the simplicity of the language used in scientific ab-
stracts. Other more elaborate methods have been attempted
for non-scientific text (e.g. Reynar and Ratnaparkhi, 1997).
The extracted sentences are scored by averaging the z-
score values of the individual words. During the analysis, it
and n is the total number of families.
becomes evident that the sentences are very useful for inter-
The dictionary of the background set of protein families,
pretation of the keywords since they provide the appropriate
and the frequency and deviation of its words to evaluate new
The algorithm has been implemented in a Web server that
Provided with a query family with an associated set of MED-
accepts text from multiple abstracts, e.g. a concatenation of
LINE abstracts, we can now evaluate the words that are like-
abstracts proceeding from a search in MEDLINE. The re-
ly to be important for the family (putative keywords) by com-
sults are given as Web pages with relative links to facilitate
information retrieval. The information provided includes
First we compute the dictionary of the words used in the
keywords, sentences and evaluated abstracts with their
MEDLINE abstracts provided for the query family. The fre-
corresponding scores. The keywords are linked to the sen-
quency of each of these words is then calculated as:
tences containing them, and the sentences themselves are
M.A.Andrade and A.Valencia
linked to the abstracts in which they were found. If the ab-
used is able to highlight significant words that are not very
stracts have a MEDLINE identifier, they will be linked to the
frequent, but differ significantly from the background, e.g.
NCBI MEDLINE. The abstracts are presented highlighted
‘heterozygote’ has the highest z-score (57.35), even though
with the more significant sentences and keywords.
its frequency is only 22%. Inversely, some very abundantwords are considered less significant, since they differ little
from the composition found in the background distribution,e.g. ‘genetic’ has a low z-score (3.53) even though its fre-
Analysis of the background distribution of
quency is 39%. It is also interesting that some words, such us
MEDLINE abstracts related to protein families
‘cancer’, given their connotation, seem to be a very attractivechoice for most authors and they end up scoring high in our
The performance of this system depends critically on the
system. With a z-score of 8.93 and a frequency of 44%,
composition of the background distribution used as a refer-
‘cancer’ has the highest frequency among the words with
ence. For this reason, we first assessed the quality and quan-
tity of the information contained in the background set de-
The most significant sentences selected by the system are
signed for the prototype system. The algorithm was applied
also shown in Figure 2. Sentences are easier to follow than
to the families used for the background distribution. The key-
single words, since they include fundamental contextual in-
words obtained for each of 71 protein families were com-
formation. A clear, descriptive sentence could be the sixth
pared with the words selected by human experts for annotat-
one in Figure 2, with an average z-score of 6.77: ‘ataxia telan-
ing the corresponding entries in the database. For the purpose
giectasia is a genetic disorder with an autosomic recessive
of this comparison, the words included in the SwissProt (Bai-
transmission’ (keywords are underlined). Many of the other
roch and Apweiler, 1997) KW and DE fields were treated in
high-scoring sentences are also informative, and a human
the same way as the words extracted from the MEDLINEabstracts: they were computed when they appeared asso-
expert would have no difficulty in choosing the most ap-
ciated to >50% of the proteins of a family and showed a z-
score > 0.10. The number and quality of both sets of key-
For the purpose of comparison, it can be said that the corre-
words were then compared by eye for each family. The re-
sponding database entry (HS24551 in EMBL) is described
sults obtained with this system and with the extraction of
as ‘human phosphatidylinositol 3-kinase homolog (ATM)
SwissProt keywords are available as additional material.
mRNA’ and ‘Ataxia-Telangiectasia mutated’, whose mean-
Our assessment is that, for 16 families, the background dis-
ing is not immediately obvious to the reader. The second in-
tribution contains more information than the equivalent
formation provided by the database entry is a pointer to a
SwissProt keywords, for 12 families the performance was
single MEDLINE reference that corresponds to a paper en-
similar, and for 31 families the background contains substan-
titled ‘A single gene with a product similar to PI-3 kinase’.
tial information, but not as much as the SwissProt entries.
It seems clear that in this example our system brings more
Only in the remaining 12 cases does the system perform quite
easily interpretable functional information.
poorly and assigns only one keyword. This is due mainly to
A second example was obtained by querying the system
the heterogeneity and small number of abstracts that are
with abstracts containing the words ‘ran’ and ‘GTPase’, re-
linked directly in SwissProt. Better results are expected for
lated in principle to the function of the small GTPase ‘ran’.
the analysis of more extensive and coherent sets of abstracts.
This protein is implicated in nuclear transport and belongs to
In any case, the background distribution contains a large
the large superfamily of ras-related proteins. This example is
and diverse set of words associated with different protein
presented to illustrate some of the performances of the cur-
families, providing an adequate reference set for the analysis
rent system, in particular, how useful the sentence analysis
of the information contained in other protein families, at least
is. This case also shows how the system is tolerant to some
at the level of the prototype system.
errors in the selection of the initial set of texts.
In this case, the words with higher z-scores were ‘binding’,
‘hydrolysis’, ‘GTPase’, ‘GTP’, ‘GDP’, ‘exchange’, ‘ras’,
Automatic annotation of protein families
‘tc4’, ‘binding’, ‘nuclear’, ‘import’. Many of these words are
In the first example, we analyze abstracts obtained by query-
clearly descriptive of the function of the protein family and
ing MEDLINE with the word ‘ataxia telangiectasia’, which
could be used directly as keywords. For example, ‘ras’ refers
refers to a human disease associated with a disorder of a par-
to the protein superfamily to which ran belongs, and ‘GTP’
ticular protein. The most significant words obtained (Figure
and ‘GDP’ are the cofactors bound by this protein family,
2) are directly related to the function of the protein and to its
while ‘binding’, ‘hydrolysis’, ‘GTPase’ and ‘exchange’
genetic origin, such us ‘recessive’, ‘disorder’, ‘atm’ (the
refer to the differentiated enzymatic activity of the protein
gene name), ‘predisposition’ to the disease, ‘heterozygote’
that uses GTP and afterwards replaces the used GDP for a
mode of ‘inheritance’. In many cases, the z-scoring scheme
new GTP. Finally, ‘nuclear ’ and ‘import’ are related to the
Automatic annotation of protein function Fig. 2. Results of the analysis of 100 abstracts containing ‘ataxia telangiectasia’. The results are shown in the original Web interface format. Words, abstracts and sentences are cross-linked, and it is possible to travel between them. (a) Keywords and the MEDLINE identifiers of the abstracts in which they were found, scored and selected by our algorithm. Five words, including ‘atm’, which is the gene name, or those used for the query, ‘ataxia’ and ‘telangiectasia’, were not present in the background distribution, but were selected due to their high frequency (>20%). A total of 21 words with z-scores > 0.20 were reported. Only some are shown in the screen dump of this figure. Best z-scoring words (with frequency indicated in percentage): ‘heterozygote’ (57.35, 22%), ‘recessive’ (29.73, 37%), ‘radiation’ (23.08, 41%), ‘disorder’ (12.30, 37%), ‘patient’ (9.79, 39%), ‘autosomal’ (9.02, 33%), ‘cancer’ (8.93, 44%), ‘familial’ (8.28, 23%). (b) Best sentences with the highlighted significant words (in bold face). Note that ‘at’ is an abbreviation of ‘ataxia telangiectasia’, as in the third sentence, and not the preposition ‘at’. (c) One of the abstracts used. Best sentences and words are highlighted (in italics and bold face, respectively).
The importance of the usage of sentence scoring schemes
is highlighted by the results obtained with sets of abstractsselected by keywords and shows the superiority of our algo-rithm to this kind of simplistic search. For example, in the setof abstracts for the ‘ran’ family, one completely unrelatedabstract was included in the analysis. This abstract containedthe word ‘ran’, in this case with the meaning ‘ran counter’. None of the sentences associated to this abstract were se-lected as relevant due to the absence of other significantwords. The system appears able to tolerate a certain amountof noise in the selection of the input set of abstracts.
A last example is chosen to illustrate the dependency of the
information contained in the literature analyzed. The resultsobtained by triggering the system with the words ‘carbonic’and ‘anhydrase’, restricted to abstracts of articles publishedduring two years (1980 and 1997), are compared in Figure3. There are some significant words common to the two sets,like ‘carbon’ or ‘CO2’, the cofactor of the protein family. Interestingly, there are some significant words that are verydifferent between the two sets. For example, ‘acetazolamide’was highly significant in the first year, but not later, and ‘ii’for a second isoform of the enzyme is only significant in re-cent years. This is because the initial studies on carbonic
cellular function of this protein, and ‘rtc4’ is the name of the
anhydrase put much emphasis on discovering inhibitors that
allow the manipulation of the enzymatic activity by ‘acetazo-
The meaning of single words becomes clearer when they
lamide’, which later became standard knowledge and was no
are embedded in the context of the corresponding sentences,
longer mentioned in the later abstracts. By the time of the
such as ‘like all ras related GTP binding proteins, gsp1p un-
second period, a second isoform had been discovered (iso-
dergoes cycles of GTP hydrolysis and GDP GTP exchange’,
form ‘ii’), triggering significant interest and a corresponding
which is the best scoring sentence (10.09). An interesting
phrase is ‘ran, a GTPase involved in nuclear processes’ (9.40,
The system depends on the input information. As in most
protein families, functional research is still in active progress
M.A.Andrade and A.Valencia
TREZ system [http://www3.ncbi.nlm.nih.gov/Entrez/in-dex.html (Schuler et al., 1996)]. The vector information isused by the NEIGHBOUR system (Wilbur and Coffee,1994) to compare individual abstracts with the backgrounddistribution of words in MEDLINE, and individual abstractsare compared to discover similarities.
Our approach attempts to extract relevant information con-
tained in the abstracts. We are therefore interested in analyz-ing the set of abstracts related to protein families rather thanin comparing individual abstracts.
Our system is based on the consideration of protein fam-
ilies (proteins related by an evolutionary link), and it evalu-ates the information contained in MEDLINE abstractsgrouped by protein families rather than segregated in indi-vidual abstracts. The discrimination of keywords from non-informative words thus occurs within and between proteinfamilies. In our system, protein families are used to build abackground distribution of words specific to the knowledge
Fig. 3. Best keywords extracted for two queries with abstracts
domain of protein functionality. The significance of word
containing ‘carbonic’ and ‘anhydrase’ in the years 1980 and 1997.
frequency in sets of abstracts is estimated by comparing them
The significant words common to both queries are bold faced andlinked. The tag ‘new’ in the z-score column indicates that a given
with a background distribution obtained from a selected set
word was not found in the background distribution, indicating its
exclusive relationship with the protein family under study.
An example can illustrate the difference between consider-
ing individual abstracts and protein family-related informa-tion. A word full of biological meaning, such as ‘membrane’,
and the extracted descriptions are necessarily a product of
can be found in many abstracts of articles describing pro-
teins. A particular abstract may have the word many times,but we cannot ascertain whether it refers to the protein func-
Discussion
tion, or to a technique (e.g. ‘membrane’ can be used either inthe context of a transmembrane protein or it can refer to a
The analysis of a set of abstracts related to protein families
dialysis membrane used to extract the protein). If we con-
is carried out comparing word frequency with background
sider protein families, the situation is clearly different. The
distributions in broad sets of protein families. The words se-
word ‘membrane’ will appear in most of the abstracts asso-
lected are found in most cases to be good indicators of differ-
ciated with some specific protein families, but very seldom
ent aspects of protein function and can be used as a guide for
in abstracts concerning others. It should be possible to con-
database annotations, sentences help to understand the func-
clude that ‘membrane’ is a keyword for some protein fam-
tion of the protein family under study in a summarized way,
and the highlighted abstracts save time during bibliographic
It is conceivable that more complex approaches would lead
searches. Three different examples have been presented to
to further improvements. Part of our intention with this first
illustrate how keywords, sentences and abstracts are selected
communication is to trigger the interest of researchers in the
area of language understanding applied to the annotation ofbiological function.
The proposed system is based on simple word statistics withtechniques similar to those used in statistical approaches to
language understanding (Jacobs, 1992; Allen, 1994; Wilburand Coffee, 1994). The difficulties are, therefore, those that
At least five important features are required to extend the
arise from the interpretation of free-style text (human-
current prototype to a fully operational system.
In the first place, correlation between words should be con-
A vectorial system for comparing text has been used in
sidered. In this case, words that do not score high themselves
other domains, and a variant based on neighbor relation has
can be discovered by their association with other words. The
been especially useful in the biological domain, where
problem with negative sentences can also be addressed
MEDLINE abstracts are already scanned by word in the EN-
through the study of short-range correlations between words. Automatic annotation of protein function
Second, a larger background distribution should replace
Casari,G. et al. (1995) Challenging times for bioinformatics. Nature,
the one derived from the 71 protein families used in this
376, 647–648.
study. They can be derived from other definitions of protein
Cowie,J. and Lehnert,W. (1996) Information extraction. Commun.
families, e.g. PROSITE (Bairoch et al., 1997), pfam (Sonn-
ACM, 39, 80–91.
hammer et al., 1997), or recently derived sets of protein
Etzold,T., Ulyanov,A. and Argos,P. (1996) SRS: information retrieval
clusters (Holm and Sander, 1998). Any of these sets include
system for molecular biology data banks. Methods Enzymol., 266,
more functional diversity than the HSSP database used here.
Third, a larger corpus of textual information should be ana-
Gaasterland,T. and Sensen,C.W. (1996) Fully automated genome
analysis that reflects user needs and preferences—a detailed
lyzed for each query. This can be achieved by scanning
introduction to the MAGPIE system architecture. Biochimie, 78,
MEDLINE abstracts for long periods of time or including
full papers rather than abstracts alone.
Guigó,R., Johansson,A. and Smith,T.F. (1991) Automatic evaluation
Fourth, the selection of sentences can be optimized to
of protein sequence functional patterns. Comput. Applic. Biosci., 7,
avoid spurious short or partial sentences by implementing a
sentence size-dependent weighting scheme.
Hobohm,U. and Sander,C. (1994) Enlarged representative set of
Fifth, the sentences selected in some cases are similar. It
protein structures. Protein Sci., 3, 522–524.
would be interesting to select sets of sentences with comple-
Holm,L. and Sander,C. (1998) Removing near-neighbour redundancy
mentary meaning. The analysis of word overlap between
from large protein sequence collections. Bioinformatics, 14,
sentences could be implemented to achieve this goal.
Unfortunately, some of these enhancements will signifi-
Jacobs,P.S. (1992) Text-based Intelligent Systems: Current Research
cantly increase computation costs. On a typical UNIX
and Practice in Information Extraction and Retrieval. Lawrence
workstation, it took 6 min to extract and analyze a protein
family of 62 abstracts against the background of 71 families.
Koonin,E.V., Tatusov,R.L. and Rudd,K.E. (1996) Protein sequence
We estimate that roughly 100 times more effort will be re-
comparison at genome scale. Methods Enzymol., 266, 295–322.
quired for a system working with a larger reference set,
Pearson,W. and Lipman,D. (1988) Improved tools for biological
hundreds of abstracts for the family to be analyzed and scor-
sequence comparison. Proc. Natl Acad. Sci. USA, 85, 2444–2448.
ing single words and pairs of words. At that point, more
Reynar,J.C. and Ratnaparkhi,A. (1997) A maximum entropy approach
sophisticated computational techniques will have to be used.
to identifying sentence boundaries. In Proceedings of the 5thConference on Applications of Natural Language Processing,Washington, DC, pp. 16–19. Acknowledgements
Salton,G. (1989) Automatic Text Processing. Addison-Wesley Series in
We thank Chris Sander for various encouraging discussions
Computer Science. Addison-Wesley, Reading, MA.
on this work. This work was supported under EC-TMR grant
Schneider,R., de Daruvar,A. and Sander,C. (1997) The HSSP database
‘GeneQuiz’. M.A.A. holds a postdoctoral fellowship from
of protein structure-sequence alignments. Nucleic Acids Res., 25,
Schuler,G.D., Epstein,J.A., Ohkawa,H. and Kans,J.A. (1996) Entrez:
molecular biology database and retrieval system. Methods Enzy-References mol., 266, 141–162.
Sonnhammer,E., Eddy,S. and Durbin,R. (1997) Pfam: a comprehen-
Allen,J. (1994) Natural Language Understanding. Benjamin/Cum-
sive database of protein domain families based on seed alignments.
Altschul,S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990)
Proteins, 28, 405–420.
Basic local alignment search tool. J. Mol. Biol., 215, 403–410.
Tamames,J., Ouzounis,C., Sander,C. and Valencia,A. (1996) Genomes
Bairoch,A. and Apweiler,R. (1997) The SWISS-PROT protein
with distinct functional composition. FEBS Lett., 389, 96–101.
sequence data bank and its new supplement TrEMBL. Nucleic Acids
Ulmschneider,J.E. and Doszkocs,T. (1983) A practical stemming
Res., 25, 31–36.
algorithm for online search assistance. Online Rev., 7, 301–315.
Bairoch,A., Bucher,P. and Hofmann,K. (1997) The PROSITE data-
Wilbur,W.J. and Coffee,L. (1994) The effectiveness of document
base, its status in 1997. Nucleic Acids Res., 25, 217–221.
neighboring in search enhancement. Inf. Process. Manage., 30,
Berry,M.W., Dumais,S.T. and O’Brien,G.W. (1995) Using linear
algebra for intelligent information retrieval. SIAM Rev., 37,
Zipf,G.K. (1935) The Psycho-Biology of Language. MIT Press (1968),
FOOT & ANKLE CLINICS OF AMERICA PATIENT REGISTRATION FORM PATIENT PERSONAL INFORMATION Last Name First Name MI __ American Indian/Alaskan Native __ Asian __ Black/African American __ Hawaiian/Pacific Islander __ White __OtherShoe Type: __ Dress __ Work Boot __ Steel/Ceramic Toed PATIENT CONTACT INFORMATION Home PATIENT HEALTH SERVICES INFORMATION Primary Care Physician Name:
Division of Targeted Research Fax +41(0)31 305 29 70 E-Mail [email protected] NRP Endocrine Disruptors Intermediate Summary Biological activity of complex mixtures of endocrine disruptors Project leader Prof. Dr. Hanspeter Naegeli Project number 4050-66572 English Summary Biological activty of complex estrogenic mixtures There is general concern that the permanent
 M.A.Andrade and A.Valencia
M.A.Andrade and A.Valencia Automatic annotation of protein function
Automatic annotation of protein function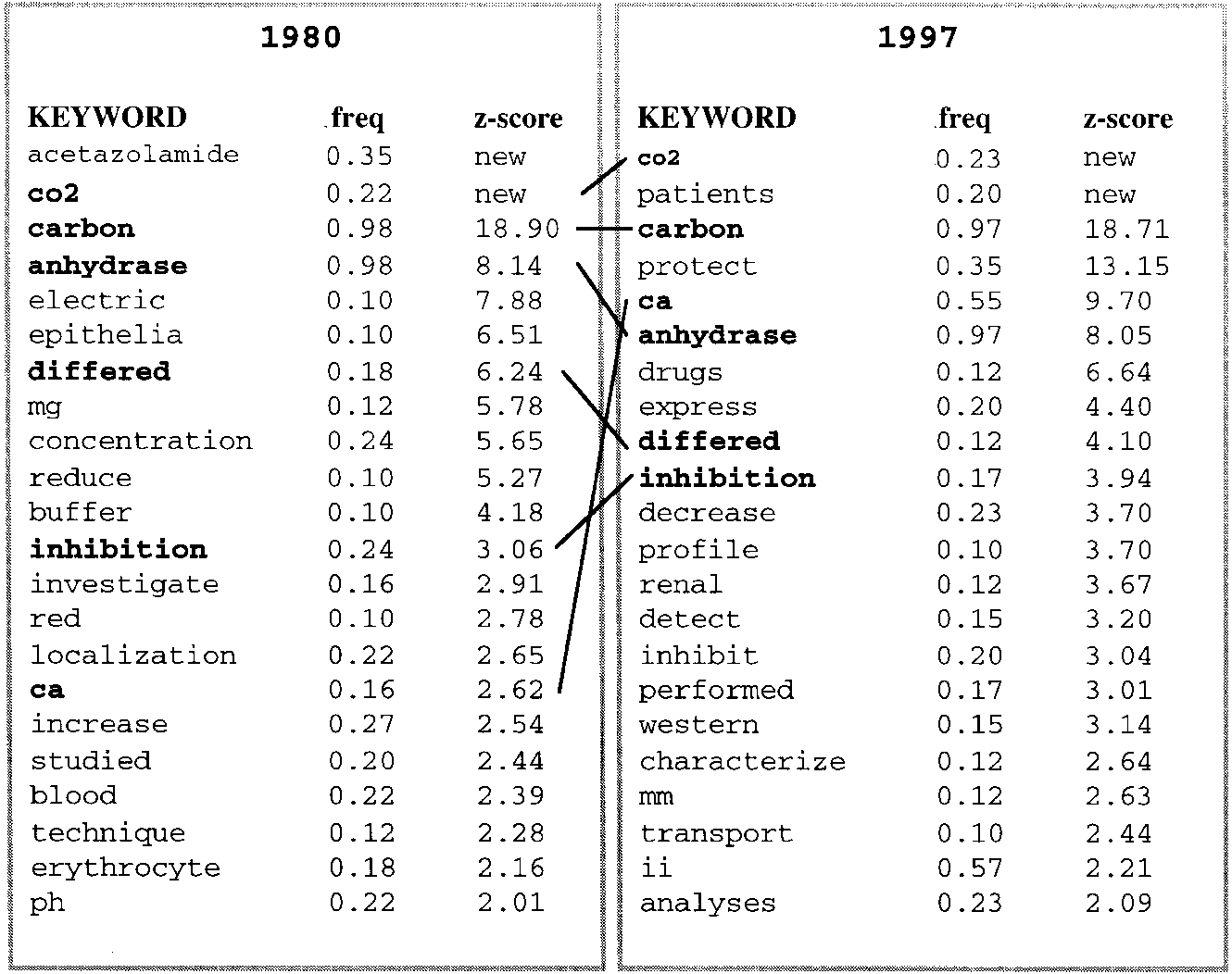 M.A.Andrade and A.Valencia
M.A.Andrade and A.Valencia