Die Struktur von Tadalafil erlaubt eine selektive Bindung an die Bindungsstelle der PDE5 und minimiert gleichzeitig die Interaktion mit PDE6, was visuelle Nebenwirkungen einschränkt. Seine Verteilung im Organismus erfolgt breit, wobei das Verteilungsvolumen etwa 63 Liter beträgt. Über 90 % des Wirkstoffs sind an Plasmaproteine gebunden. Die Wirkung bleibt unabhängig von der Nahrungsaufnahme konstant. Der Abbauweg über CYP3A4 kann durch Hemmer wie Ritonavir oder Ketoconazol verlangsamt werden, was die Plasmakonzentrationen deutlich erhöht. In diesem Kontext wird cialis 20mg preis häufig in Bezug auf pharmakokinetische Wechselwirkungen erwähnt.
Biolchem.ucla.edu
ã 2000 Macmil an Publishers Ltd All rights reserved 0950 ±9232/00 $15.00
From oncogene to drug: development of small molecule tyrosine kinase
inhibitors as anti-tumor and anti-angiogenic agents
1P®zer Global R&D, Groton, Connecticut, CT 06340, USA
The con¯uence of two distinct but related activities in the
accomplished to date to understand the molecular
past 10 years has dramatically accelerated eorts
pharmacology of small molecule inhibitors of receptor
towards the discovery and development of novel drugs
tyrosine kinases (Sedlacek, 2000; Fry, 2000; Bridges,
to treat cancer. The ®rst is a rapidly emerging
1999; Levitzki, 1999; Lawrence and Niu, 1998). With-
understanding that a number of distinct tyrosine kinases
out summarizing each of these important reviews, they
play roles in diverse but fundamentally important aspects
provide an appropriate context for understanding the
of tumor progression (growth, survival, metastasis and
obstacles and triumphs that have led, very recently, to
angiogenesis). The second is the discovery that small
the ®rst reproducible, objective clinical responses in
molecule compounds have the capacity to potently and
cancer patients treated with tyrosine kinase inhibitors.
selectively inhibit the biochemical function of tyrosine
The catalytic function of protein tyrosine kinases
kinases by competing for ATP binding at the enzyme
involves the simple transfer of the gamma phosphate of
catalytic site. These observations have been conjoined in
ATP to hydroxyl group of a tyrosine residue of
major eorts to bring forward into clinical development
proteins (or peptides) encompassing a diversity of
novel cancer drugs with the potential to provide both
primary sequences and tertiary structures (Songyang
clinical ecacy and improved tolerability. The focus of
and Cantley, 1998). Each of the substrates in the
this review is on the development of small molecule
phosphotransfer reaction, the tyrosine hydroxy group
tyrosine kinase inhibitors, and does not extend to other
and ATP, represent reasonable pharmacological start-
approaches that could be applied to disrupt the same
ing points for the design of substrate analogs and
pathways in clinical tumors (receptor and/or ligand-
competitive inhibitors of tyrosine kinases. A diverse set
competitive antibodies, intrabodies, antisense ribonucleo-
of pharmacophores, including natural products (laven-
tides, ribozymes, phosphatase inhibitors or SH2/SH3-
dustins and erbstatins) and synthetic tyrosine mimetics,
directed agents). Selected tyrosine kinase inhibitors,
have all been characterized on the basis of their ability
known or believed to be in development in cancer
to competitively inhibit tyrosine kinase function
treatment trials, are summarized as are some of the
(Levitzki, 1999). These compounds tended to have
key issues that must be addressed if these compounds are
poor potency (particularly in cells), to yield relatively
to be developed into clinically useful cancer chemother-
¯at structure-activity relationships, and to be some-
apeutic agents. Oncogene (2000) 19, 6574 ± 6583.
what non-speci®c in their kinase inhibition (Fry, 2000).
Attacking this reaction from the other side, by
Keywords: tyrosine kinase inhibitors; anti-tumor; anti-
identifying compounds that mimic ATP, was originally
thought to be even less tractable. As reviewed by
Lawrence and Niu (1998), the theoretical obstacles
were immense. First, the primary sequence of the ATP-
Origin of species ± brief overview of substrate-based
binding pocket of all kinases is highly conserved, and
therefore selectivity, if not speci®city, represents a
signi®cant technical challenge. Secondly, the intracel-
Among all non-traditional (non-DNA-directed) cancer
lular concentration of ATP can exceed 5 mM,
targets for which pharmacological intervention is
particularly in tumor cells, while the Km for ATP in
feasible, there are none that have generated as much
most kinase active sites is in the micromolar range,
widespread interest, and have invoked as much
thus ensuring full-time saturation by ATP. ATP-
resource investment in both the public and private
competitive inhibitors would need to exhibit at least
sectors in the past 7 years, as have the tyrosine kinases.
nanomolar inhibitory kinetic constants to eectively
Several excellent recent reviews have described the
compete in this circumstance (Lawrence and Niu,
functions of various tyrosine kinases in the key
1998). Finally, there are multiple non-kinase ATP-
pathways that drive tumor progression, from ®rst
dependent enzymes important to normal physiology,
genetic insult to disseminated disease (Hanahan and
and so an indiscriminant ATP mimetic would likely
Weinberg, 2000; Hunter, 2000; Gibbs, 2000). Key
have toxicities that were pharmacologically and
among these are the receptor tyrosine kinases which
initiate signal transduction in tumor cells or endothelial
This theoretical logjam was broken in convincing
cells following the binding of the growth factors EGF,
fashion when the tyrosine kinase inhibitory activities of
PDGF and VEGF. There are also several excellent
anilinoquinazolines were ®rst described in 1994 by
reviews that provide detailed overviews of the work
three separate groups (Fry et al., 1994; Ward et al.,
1994; Osherov and Levitzki, 1994). For example, the
work of Fry et al. (1994) at Warner Lambert revealed
that 4-anilinoquinazolines were potent (nM) inhibitors
Tyrosine kinase inhibitors in cancer treatment trials
of the EGFR tyrosine kinase with good cell activity
relapsed on drug between 45 and 81 days. Of 19
and profound biochemical selectivity relative to other
responding patients, 10 experienced Grade 3 ± 4 neu-
kinases within the tyrosine kinase family. Further
tropenia. This response rate, and the incidence of
elaboration of structure-activity relationships rich in
Grade 3 ± 4 toxicity, compares very favorably to the
new possibilities resulted in ATP-competitive inhibitors
standard of care cytotoxic chemotherapies for CML.
of the EGFR tyrosine kinase with Ki values in the
As such, more de®nitive trials assessing the ecacy and
single digit picomolar range. It is interesting to note
safety of STI 571 are ongoing in CML.
that the Michaelis-Menten equation could not be used
It is interesting to speculate as to the biochemical
to derive the Ki values of these molecules. So avid was
basis for both the ecacy and the toleration pro®le of
the binding of compound to the ATP site, the
STI 571. Two other tyrosine kinases potently inhibited
conventional approximation that total and free enzyme
by STI 571, c-kit and PDGFR, are both believed to play
concentrations were equivalent did not apply under
important roles in maintaining bone marrow stroma ±
these conditions. These accomplishments, which may
progenitor cell interactions (Ashman, 1999; Sungaran et
be among the most important in pharmacology for the
al., 2000). Thus, inhibition of c-kit and PDGFR could
last 10 years, were largely achieved by empirical
also account for some of the compelling clinical activity
screening and iterative medicinal chemistry. Even more
of STI 571 in CML, as well as for its toxicity pro®le
new chemotypes may emerge as structure-based design
(neutropenia). Treatment of a c-kit expressing a human
becomes more commonly applied to the identi®cation
myeloid leukemia cell line, M-07e, with STI 571 before
of both active site- and allosteric site-directed inhibitors
stimulation with kit ligand inhibited c-kit autopho-
for an ever-widening slate of tyrosine kinase targets.
sphorylation, activation of mitogen-activated protein
While these early lead molecules had biopharmaceu-
(MAP) kinase, and activation of Akt, with an IC50 of
tical properties which were by-and-large incompatible
100 nM (Heinrich et al., 2000). STI 571 was even more
with oral bioavailability and good duration of exposure
potent in a human mast cell leukemia cell line (HMC-1)
in vivo, the results spurred on a number of groups,
expressing an activated mutant form of c-kit. Similar
which have since identi®ed and developed tyrosine
results have also recently been reported in non-
kinase inhibitors with signi®cant potential to treat
hematopoietic tumor cells (Wang et al., 2000). The
ecacy and safety hypotheses for inhibition of c-abl in
CML may perhaps only be addressed with a more
selective abl tyrosine kinase inhibitor. Given the
Selected development candidates ± updates
apparent therapeutic bene®t of STI 571, this may be
largely an academic question, but one with important
implications as one tries to rationalize the desired
selectivity pro®les of tyrosine kinase inhibitors most
STI 571 (CGP57148B) Among all of the candidates
likely to generate both ecacy and safety in humans.
currently in clinical development, perhaps none has
provided as much `proof of concept' for the clinical
SU101 (le¯unomide; HWA 486) Le¯unomide was
ecacy and tolerability of small molecule tyrosine
originally described and developed as an inhibitor of
kinase inhibitors as has STI 571. Originally disclosed by
dihydroorotate dehydrogenase, a key enzyme in the de
Novartis as a multitrophic tyrosine kinase inhibitor,
novo synthesis of pyrimidines, for use as an immuno-
STI 571 was described by Druker et al. (1996); and
suppressive or anti-arthritic agent (Bartlett and
Druker and Lydon (2000) as having potent activity vs
Schleyerbach, 1985; Kuo et al., 1996). Le¯unomide
the translocation product bcr-abl, the transforming
has shown signi®cant activity as a treatment for
tyrosine kinase found in virtually all CML cells
rheumatoid arthritis (Smolen and Emery, 2000; Cohen
expressing the Philadelphia chromosome (Kurzrock et
et al., 2000b), and was launched by Aventis as Arava2
al., 1988; Kelliher et al., 1990). The inhibition of v-abl,
in the US and elsewhere beginning in 1998. Extending
bcr-abl and PDGFR autophosphorylation by the 2-
the work of others (Mattar et al., 1993; Xu et al.,
1995), Shawver and co-workers reported that micro-
lar concentrations was found to translate to both in
molar concentrations of le¯unomide inhibited the
vivo anti-tumor activity, and to the inhibition of
autophosphorylation of the tyrosine kinase receptors
clonogenicity of blasts from CML patients (le Coutre
for PDGF and VEGF (Shawver et al., 1997). The
et al., 1999; Druker et al., 1996). The results of a
compound was also eective at blocking mitogenesis
clinical trial in which STI 571 was administered to
stimulated by both PDGF and EGF, but exogenous
CML and ALL patients expressing bcr-abl in their
uridine could not reverse the eect of le¯unomide on
leukemic blasts were most recently summarized in May
PDGF mitogenesis, suggesting that inhibition of the
2000 (Talpaz et al., 2000). STI 571 was used to treat 33
receptor tyrosine kinase, and not inhibition of
acute leukemia patients, which included 21 myeloid
pyrimidine pools, was a key pharmacological activity.
blast crisis CML patients and 12 bcr-abl-positive ALL
The inhibition of EGF-induced mitogenesis by le¯u-
or lymphoid blast crisis CML patients. Clinical
nomide was reversed in part by uridine (Shawver et al.,
responses, as de®ned by a decrease in the percentage
1997), despite the fact that le¯unomide and close-in
of patients achieving reduction in bone marrow blasts
analogs also have inhibitory activity vs the EGFR
to 15% of pre-treatment levels, were observed in 55%
tyrosine kinase (Ghosh et al., 1999).
of myeloid blast crisis patients, with complete responses
Le¯unomide/SU101 is clearly a tyrosine kinase
in 22% of these patients. The response rates in patients
inhibitor with multiple biochemical eects, and readily
with bcr-abl positive ALL and lymphoid blast crisis of
generates a predominant active metabolite (SU0020 or
CML were higher (82% with 55% complete responses),
A771726; Figure 1) that has a complex inhibitory
but all of the patients with lymphoid leukemias
pro®le of its own (Hamilton et al., 1999). SU 101 was,
Tyrosine kinase inhibitors in cancer treatment trials
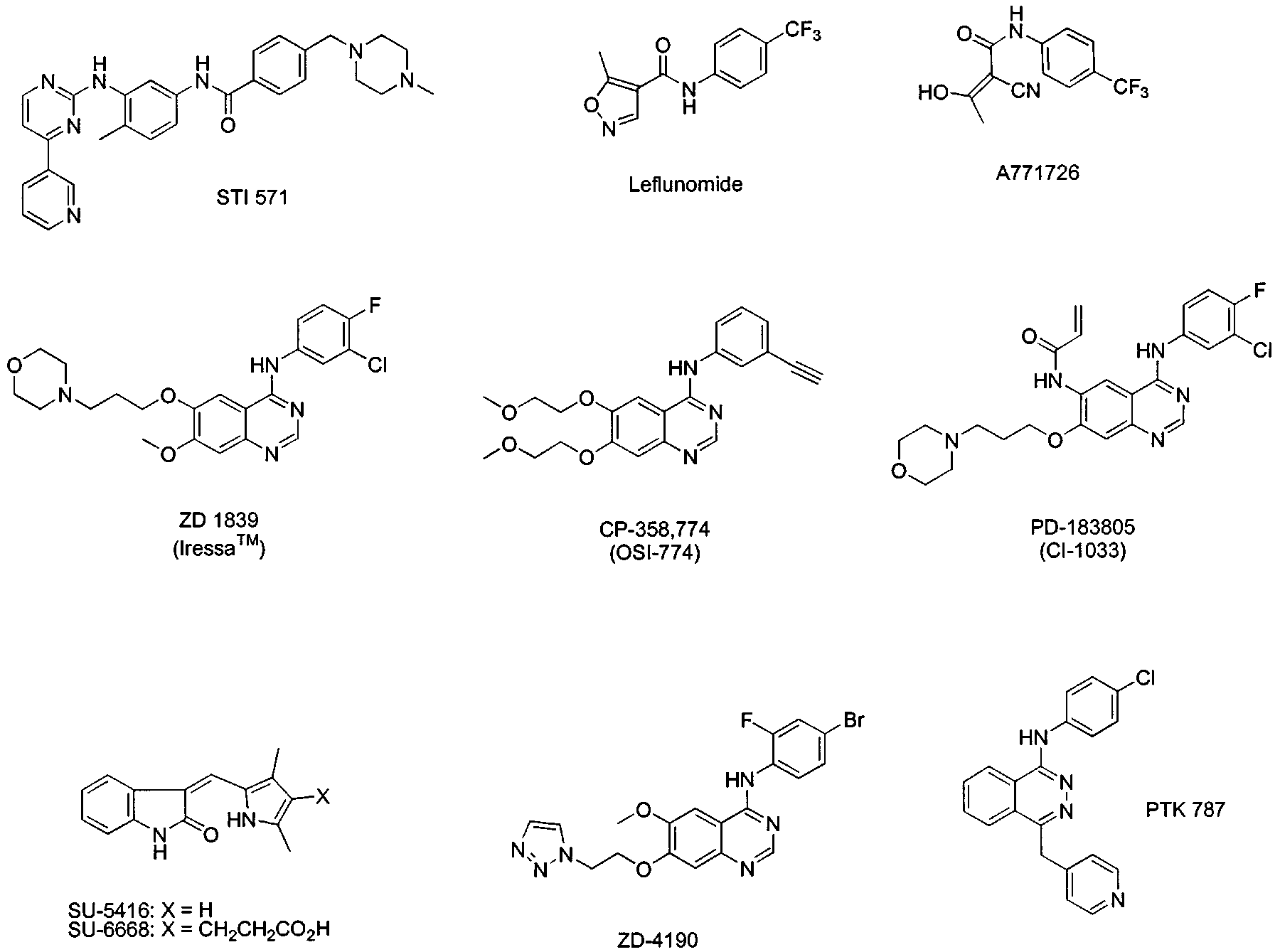
Figure 1 Structures of selected tyrosine kinase inhibitors in clinical development for cancer
nonetheless, progressed into clinical trials by SUGEN
clinical ®ndings with AstraZeneca's ZD1839 (Iressa2)
(now part of Pharmacia). A Phase I study in cancer
have been equally compelling. The pharmacological
patients revealed that SU 101 was well-tolerated as a
characteristics of Iressa2 were ®rst described in 1996
24 h continuous i.v. infusion at doses up to 443 mg/m2/
(Wakeling et al., 1996; Woodburn et al., 1997) as a
wk. At this dose, the plasma concentration of the
potent and selective inhibitor of the EGFR tyrosine
active metabolite was maintained at levels sucient to
kinase. This quinazoline-based compound (Figure 1) is
block both PDGFR and EGFR signaling, as well as
an ATP-competitive inhibitor of the EGFR tyrosine
pyrimidine biosynthesis (Eckhardt et al., 1999). Toxi-
kinase (IC50 25 nM) with 50-fold selectivity relative to
cities were relatively minor (Grade 1 ± 2 nausea,
closely homologous erbB family members (IC50 for
vomiting and fever in approximately 20% of all
erbB2 1 ± 3 mM) and even greater selectivity for more
courses given). Surprisingly, hematopoietic toxicities
divergent tyrosine kinases. It demonstrates good
and hemolysis, which had been noted in the preclinical
cellular potency (80 nM IC50 for inhibition of EGF-
experience with SU 101, were not seen in this Phase I
dependent mitogenesis) and robust, dose-dependent
population. One partial response was seen in 26
anti-tumor ecacy in a variety of human tumor
patients receiving an average of two courses each; the
xenografts (Woodburn et al., 1997). These results have
responding patient received 13 courses (52 infusions) to
been most recently extended to show that Iressa2 has
treat an anaplastic astrocytoma, and had a notable
in vivo ecacy in a diverse human tumor xenograft
(450%) reduction in one measurable lesion (Eckhardt
models both with (Ciardello et al., 2000) and without
et al., 1999). SU 101 has been reported to be in
(Sirotnak et al., 2000) highly activated EGFR signaling
advanced trials for multiple solid tumor types, but
pathways. Of equal interest are the observations that
recent disclosures (Garber, 2000) indicate that Phase
Iressa2 combines with standard cytotoxic agents
III trials in at least one tumor type (glioblastoma) have
(platinums, taxanes, topoisomerase I inhibitors, etc.)
been abandoned. The status of other trials (ongoing
to produce additive or supra-additive anti-tumor
Phase II trials for ovarian and NSCLC; planned Phase
ecacy in vivo without exacerbation of the toxicity of
III trials for prostate, colon and NSCLC) is uncertain
the co-administered cytotoxics. The ®ndings that tumor
EGFR density does not predict ecacy when the
compound is used in conjunction with cytotoxic agents
have signi®cantly impacted the development strategy
employed by AstraZeneca as Iressa2 moves towards
Iressa2 (ZD1839) While STI 571 has provided no-
Multiple Phase I trials with Iressa2 have been
table clinical proof-of-concept for the clinical ecacy
summarized, and the results revealed reasonable
and safety of tyrosine kinase inhibitors, the early
pharmacokinetics, good toleration and the ®rst signs
Tyrosine kinase inhibitors in cancer treatment trials
of clinical ecacy when used as a single agent in
(Pollack et al., 1999; Goss et al., 2000; Allen et al.,
patients with advanced disease (Ferry et al., 2000;
2000). Given the overall safety and toleration pro®le of
Baselga et al., 2000; Kelly et al., 2000). Following oral
Iressa2, AstraZeneca has committed to an aggressive
administration of a single dose (50 mg), maximum
development strategy, which includes two large Phase
plasma drug concentrations (mean 45 ng/ml) occurred
III studies to assess the use of Iressa2 in combination
1 ± 5 h post-dose. The mean terminal t1/2 was 34 h.
with cis- or carbo-platinum plus a taxane or
Inter-subject variability in exposure was signi®cant
gemcitabine in ®rst-line therapy for NSCLC (trials 14
following single and multiple administration (up to
and 17), as well as a Phase II trial (trial 16) to con®rm
sevenfold at each dose level), but exposure increased
the single agent activity of Iressa2 in patients with
proportionally with dose, with no apparent change in
advanced NSCLC (Kelly et al., 2000). It is important
terminal t1/2 across the dose range tested (Kelly et al.,
to note that these trials do not call for a prospective
2000). In a larger dose-escalation trial, Ferry and
selection for patients with tumors with some pre-
collaborators administered Iressa2 at doses of 50 ±
de®ned level of EGFR over-expression. All epithelial
700 mg once daily, given orally for 14 days followed by
tumors express some EGFR, and in the disease target
14 days of observation (Ferry et al., 2000). In total, 64
here, NSCLC, tumors often present with a high
patients with advanced disease, who had each
proportion of EGFR over-expression (up to 80 ± 90%
progressed while on prior chemotherapy, completed
in advanced disease). The strategy is also consistent
145 cycles. Cmax and AUC0-24h were proportional
with pre-clinical data suggesting that ecacy in drug
across the entire dose range (mean values 113 ±
combinations may not be determined in large part by
2255 ng/ml and 1.8 ± 38.5 mg.h/ml, respectively). As
the level of EGFR over-expression in tumors (Sirotnak
in single dose studies, Iressa2 showed a long terminal
et al., 2000). Results are expected from these pivotal
elimination half-life (mean of 46 h). Iressa2 was very
trials in a late-2001 or early-2002 timeframe.
well-tolerated in this study; the most common adverse
events were diarrhea and acne-like skin rash (Grade 1 ±
OSI-774 (CP-358,774) CP-358,774 is also a potent
2). Acne-like skin rashes have emerged as a common,
and selective quinazoline-based inhibitor of the EGFR
mechanism-based adverse event for EGFR inhibitors,
function (Figure 1). This compound is a reversible,
but the speci®c toxicological eect in the skin is not yet
ATP-competitive inhibitor (IC50 of 2 nM) of the EGFR
well understood. Grade 3 ± 4 adverse events were
tyrosine kinase, with greater than 500-fold selectivity
shown to be rare with Iressa2 treatment, and were
against other tyrosine kinases, such as the closely
generally ascribed to disease progression. The dose-
related erbB2 kinase, as well as v-src, c-abl and the
limiting toxicity, de®ned at the 700 mg dose level, was
insulin and IGF-1 receptors, (Moyer et al., 1997). CP-
Grade 3 diarrhea (Ferry et al., 2000).
358,774 inhibits the autophosphorylation of the EGF
A compelling level of ecacy was also revealed in
receptor in a variety of EGFR over-expressing tumor
these early trials (Ferry et al., 2000). Anti-tumor
cells (IC50=20 nM), and produces cell cycle arrest and
responses were most evident among the 16 NSCLC
apoptosis in multiple cell types (Moyer et al., 1997;
patients treated with Iressa2 ± two had an objective
Barbacci et al., 1997; Iwata et al., 1997). In vivo, CP-
partial response, two patients had signi®cant regression
358,774 eectively inhibits EGFR-speci®c tyrosine
of disease and two patients had stable disease. Similar
phosphorylation in human tumor xenografts (ED50 of
pharmacokinetic and safety pro®les were noted in a
10 mg/kg p.o. when given as a single dose) with
separate study (Baselga et al., 2000), one that also
signi®cant duration of action; daily dosing produces
revealed the potential for ecacy from Iressa2 in
substantial growth inhibition and regressions in human
patients with advanced prostatic and head-neck
tumor xenografts (Pollack et al., 1999). Moreover, the
cancers. These early results added importantly to the
dose-response for tumor growth inhibition shows good
proof-of-concept that selective tyrosine kinase inhibi-
agreement with the dose-response for inhibition of
tors could have signi®cant single agent ecacy, as
EGFR-phosphotyrosine in tumors from treated ani-
measured by objective tumor regressions, in patients
mals. As with Iressa2, CP-358,774 was found to
with advanced disease. The clinical observations have
generate additive anti-tumor activity when used in
therefore recapitulated the pre-clinical data showing
combination with cis-platinum and other cytotoxic
that Iressa2 increased apoptosis and regressions in
agents, without exacerbating the toxicities of the other
human tumor xenograft models (Ciardello et al., 2000).
chemotherapeutants (Pollack et al., 1999).
The Iressa2 data indicate that the ecacy of these
Clinical studies with CP-358,774 have revealed that
agents can be measured using more classically de®ned
the agent is well-tolerated at oral doses that achieve
clinical endpoints. There will undoubtedly be signi®-
plasma concentrations projected to be required for
cant value in the use of pharmacodynamic and
anti-tumor ecacy in humans (400 ± 500 ng/ml). In one
surrogate endpoints to guide dose-intensi®cation or to
study, escalating doses were administered orally once
pre-select patients for whom other tyrosine kinase
every week (Karp et al., 1999). Eighteen patients with
inhibitors might represent the most promising treat-
advanced solid tumors were treated at ®ve doses (100 ±
ment option. Pharmacodynamic endpoints have not
1000 mg) for a maximum period of 24 weeks.
played a major role in the early development of EGFR
Toxicities were observed only at doses higher than
tyrosine kinase inhibitors, despite the fact that several
200 mg/week, and included mild fatigue, Grade 2
reasonable options exist, including both invasive
maculopapular (acneiform) rash, Grade 2 nausea, and
techniques (direct measurement of tumor-derived or
Grade 2 diarrhea. Like Iressa2, CP-358,774 exhibited
normal tissue-derived EGFR phosphotyrosine, phos-
intra- and inter-subject variability in exposure, but
phorylation of down-stream signaling molecules;
dose-proportional increases in exposure were observed
apoptosis markers) and non-invasive techniques such
throughout the 100 ± 1000 mg weekly dose range.
as PET imaging of metabolically modulated tumors
During the ®rst 24 h following a single dose, the Cavg
Tyrosine kinase inhibitors in cancer treatment trials
(0.9 ± 4.8 mg/ml for 100 ± 1000 mg doses, respectively)
has taken over complete responsibility for the devel-
was some two- to 10-fold above the projected
opment of CP-358,774, which is now formally referred
ecacious plasma concentration. No maximally toler-
ated dose or dose-limiting toxicity was discerned in this
study. In a second Phase I study (Siu et al., 1999),
CI-1033 (PD183805) As described above, the selec-
patients were given CP-358,774 tablets in a variety of
tive and reversible inhibitors of the EGFR tyrosine
dose schedules, culminating in daily dosing at the
kinase appear to oer the promise of therapeutic
maximally tolerated dose. The target Cavg of 400 ±
ecacy coupled to reasonable tolerability. It is
500 ng/ml was achievable at doses at and above
important to note, however, that the therapeutic index
100 mg/day on a well-tolerated schedule (Cavg values
of neither Iressa2 nor CP-358,774 has yet to be fully
following continuous daily dosing at the 50, 100 and
elaborated, and that there may be signi®cant proximity
200 mg/day levels were 432, 973 and 2120 ng/ml,
between the maximally tolerated doses and the
respectively). Dose-limiting diarrhea was encountered
ecacious doses for both agents. Moreover, the
at the 200 mg/day level. An intermediate dose of
ecacy of neither agent has yet to be established in a
150 mg/day was subsequently de®ned as the maximally
blinded, placebo controlled study. As such, there
tolerated dose (two of three patients had Grade 1
continues to be an opportunity to discover and develop
distinctly dierent EGFR tyrosine kinase inhibitors
Siu and co-workers also made eorts to understand
with even greater potential for ecacy and a broader
the `characteristic' Grade 1 ± 2 acneiform rash seen in
spectrum of activity. CI-1033 is one such distinctly
patients treated with CP-358,774, which was limited to
dierent development candidate. As recently reviewed
regions of the upper body where adolescent acne is
by David Fry of the former Warner Lambert
usually manifest (face, back and scalp). Histopathology
organization, signaling through the erbB family of
of skin biopsies showed subepidermal neutrophilic
tyrosine kinase receptors often involves complex cross-
in®ltration and epidermal hyperproliferation (Siu et
talk among the members of that receptor family (Fry,
al., 1999). While the precise cytopathic basis for the
2000). The four family members (EGFR or erbB;
acneiform rash has not yet been determined, the
erbB2, erbB3 and erbB4) are known to intensify their
consistent clinical observations with three dierent
kinase-dependent transforming signals via the forma-
agents targeting EGFR function (CP-358,774, Iressa2
tion of heterodimers with each other (Tzahar et al.,
and Imclone's C-225 antibody) suggest that this is a
1996). There is, therefore, a compelling rationale to
mechanism-based ®nding (Siu et al., 1999; Ferry et al.,
consider the potential utility of nonspeci®c but selective
2000; Cohen et al., 2000b). Skin changes are consis-
inhibitors that eectively block the function of the erbB
tently noted in preclinical studies with rodents exposed
family but do not inhibit more structurally diverse
to CP-358,774 for extended dosing periods, and these
toxicological results are analogous to the skin changes
There is also a strong rationale to consider
seen in the waved-2 mouse, which has a mutated and
irreversible tyrosine kinase inhibitors. The reversible
marginally functional EGFR tyrosine kinase (Luetteke
inhibitors have apparently generated clinical ecacy
with dosing regimens designed to maintain plasma
Early ecacy readouts from ongoing Phase II
concentrations at fairly high levels for extended periods
clinical trials with CP-358,774 have been compelling.
of time. The optimal dosing paradigm for an
The agent appears to have a broad potential to treat a
irreversible inhibitor would be less likely to require
variety of human solid tumors, including NSCLC,
prolonged exposure. Moreover, the `absolute ®nality'
breast, ovarian and squamous head and neck tumors
(Fry, 2000) of the irreversible inhibitors could con-
(Bonomi et al., 2000; Allen et al., 2000; Siu et al., 2000;
ceivably provide signi®cant advantages in terms of
Hammond et al., 2000). For example, in 34 NSCLC
antitumor ecacy. To be balanced, a multi-tropic and
patients who had failed prior chemotherapy, daily oral
irreversible inhibitor would also have the potential to
doses of 150 mg CP-358,774 were well-tolerated, with a
generate a toxicity pro®le that was dierent and,
maculopapular (acneiform) rash being the most
perhaps, without advantages relative to the more
common adverse event reported. In 56 total patients
selective, reversible inhibitors. Preclinical data suggest
evaluable for tumor response, there have been six
that irreversible EGFR tyrosine kinase inhibitors can
partial responses in the lung and/or liver at 8 weeks
generate signi®cant ecacy with good toleration
and several patients with stable disease (Bonomi et al.,
(Vincent et al., 1999), but the ultimate utility of these
2000). In 71 patients with refractory squamous
agents can only be determined in clinical trials.
carcinomas of the head and neck, CP-358,774 was
Homology modeling of ATP binding to the pocket
again found to cause a reversible acneiform rash and
of EGFR suggested that the thiol of cys773 would be a
Grade 1 ± 2 diarrhea. Of 78 patients evaluable for
key potential site for attack by a rationally designed
response, there have been at least eight con®rmed
irreversible ATP-mimetic. One compound containing
partial responses and 23 patients with stable disease
an acrylamide functionality at the six position of the 4-
(Siu et al., 2000). These preliminary results indicate
anilinoquinazoline nucleus (Figure 1) was found to
that CP-358,774 is generally well-tolerated and demon-
have a profoundly rapid onset and long-lasting
strates evidence of single agent anti-tumor activity in
inhibition of both EGFR and erbB2 in tumor cells,
patients with recurrent head and neck cancer, as well
and to be selective relative to non-erbB tyrosine kinases
(Fry et al., 1998). When compared to very closely
Due to signi®cant interests in both CP-358,774 and
related reversible analogs (in which the acrylamide
CI-1033, P®zer was directed to divest one of these two
double bond was reduced), the 6-substituted irrever-
agents as a condition of their acquisition of Warner
sible analogs were more potent in vitro and had
Lambert in 2000. As such, Oncogene Science (OSIP)
signi®cantly greater ecacy in vivo. Further improve-
Tyrosine kinase inhibitors in cancer treatment trials
ments (addition of substitutions which also improved
of FGFR, which occurred at SU 5416 concentrations
water-solubility) led to the elaboration of PD 183805/
some 100-fold higher, was found in kinetic experiments
CI-1033 (Figure 1). Like its predecessors, this com-
to be `mixed' competitive and non-competitive (Mendel
pound has excellent (low nM) potency against erbB2
et al., 2000). It has been speculated that the latter result
and EGFR in both enzyme- and cell-based assays
is due to speci®c biopharmaceutical properties of the
(Sherwood et al., 1999). Consistent with a predicted
compound, which is both lipophilic and potentially
advantage relative to reversible inhibitors, CI-1033
reactive in nature. Consistent with this concept are
potently inhibits human tumor xenografts when dosed
preliminary observations that the inhibition of VEGF-
as infrequently as once per week, and a single dose
dependent endothelial cell proliferation by SU 5416 has
eliminated the level of EGFR phosphorylation in
both a rapid onset and a pseudo-irreversible behavior
tumors for longer than 72 h (Vincent et al., 1999).
which may be due to high intracellular levels of
Like CP-358,774, CI-1033 combines well in drug
compound (Mendel et al., 2000). Inhibition of
combinations with cytotoxic agents. Given 24 h after
endothelial cell proliferation translated to anti-tumor
gemcitabine, CI-1033 produced a signi®cant increase in
ecacy in a number of human xenograft and rodent
the apoptotic fraction in tumors over treatment with
tumor models (Fong et al., 1999). In these studies, no
either drug alone (Nelson and Fry, 2000). CI-1033 also
data were generated to relate drug exposure (said to be
eectively decreased the clonogenicity of human tumor
very short-lived in rodents), or biochemical inhibition
cells taken from patients (Medina et al., 2000), with
of VEGFR or PDGFR, to anti-tumor ecacy.
notable responses seen in breast (67%), NSCLC (60%)
Interestingly, the ecacy of SU 5416 was found to be
and ovarian cancer specimens. CI-1033 Phase I clinical
greater in slower-growing vs faster growing solid tumor
trials have recently been initiated, but data on
xenografts, which led Fong et al. (1999) to speculate
pharmacokinetics or safety have not yet been disclosed.
that SU 5416 might bind preferentially to resting vs
activated tyrosine kinases on endothelial cells. This
Small molecule tyrosine kinase inhibitors targeting
would be at odds with other data suggesting that
quinazolines bind more avidly to activated kinases
(Levitzki and Bohmer, 1998) but, if true, may bode
There are multiple tyrosine kinase receptors which
well for human ecacy in a majority of clinical
appear to have key roles in the generation of new
tumor blood vessels and, as such, represent reasonable
Phase I studies were carried out in 69 advanced
targets for cancer chemotherapy (for excellent recent
disease patients, with SU 5416 dosed i.v. twice weekly.
reviews, see Cherrington et al., 2000; Randal, 2000;
Patients were treated at 13 dose levels between 4.4 ±
Thompson et al., 1999; Hamby and Showalter, 1999).
190 mg/m2/day; at the highest dose, a dose limiting
Included among the key tyrosine kinase targets that
toxicity (projectile vomiting) was observed (Rosen et
have generated the most interest in the scienti®c and
al., 1999). Induction of metabolism was noted in all
patent (Connell, 2000) literature are PDGFR, VEGFR,
patients, either due to the parent drug, a metabolite or
FGFR and tie-2. The key development candidates
dexamethasone premedication, and the elimination
targeting PDGFR, STI 571 and SU101, were described
half-life was found to be 55 min (Cropp et al., 1999).
above, though neither compound is likely to reveal the
Early signs of ecacy were also apparent, with
clinical utility of PDGFR-directed inhibition of
objective responses seen in three patients (Kaposi's
angiogenesis due to their multiple mechanisms of
sarcoma, metastatic basal cell and colorectal cancer);
action. Agents that selectively target FGFR and tie-2
seven patients remained on study for more than 6
are not known to be in development, though several
months, while two remained on study for greater than
drugs targeting VEGFR have inhibitory activity vs
18 months (Rosen et al., 1999; Mendel et al., 2000).
FGFR. As such, the focus of the remainder of this
Given these results, SU 5416 has been advanced into
overview will be on the clinical candidates targeting
multiple Phase II and III at an initial recommended
VEGFR. Two high anity receptors for VEGF have
dose of 145 mg/m2, which is sucient to produce
been identi®ed and characterized on human endothelial
systemic exposure comparable to what was required to
cells, ¯t-1 and KDR. KDR appears to be expressed
yield eective tumor growth inhibition in animals
primarily on activated endothelial cells and is thought
(Cropp et al., 1999). This dose is also within 30% of
to be more of a key driver of mitogenic responses
the human maximally tolerated dose (190 mg/m2). The
commonly found in neovascularizing tumors, while ¯t-
ongoing development plan includes large studies in
1 is expressed on multiple other cell types (Plate et al.,
NSCLC and colorectal cancer to assess the ecacy of
1994; Wedge et al., 2000a). For the purposes of this
SU 5416 both as a single agent and in combination
review, the terms KDR and VEGFR will be used
with standard chemotherapies (Mendel et al., 2000).
interchangeably, unless otherwise speci®ed.
A related agent in development, SU 6668 (Figure 1),
combines a less selective inhibitory pro®le (inhibition
SU 5416 and SU 6668 The former SUGEN organiza-
of FGFR in addition to PDGFR and VEGFR) with a
tion (now part of Pharmacia) has clearly set the early
more favorable biopharmaceutical pro®le (Laird et al.,
pace in the race to identify and develop inhibitors of
2000). SU 6668 has a signi®cantly lower Ki for PDGFR
the VEGFR tyrosine kinase. Eorts towards this end
relative to VEGFR or FGFR (8 nM vs 2.1 and 1.2 mM,
have initially focused on the indolin-2-one pharmaco-
respectively), a result which appeared consistent with
phore (Figure 1). Among the earliest compounds of
homology models of the respective active sites, but
this class was SU 5416, which was found to be a potent
inconsistent with the cellular eects of SU 6668
inhibitor of the kinase activities of both VEGFR and
(VEGFR-stimulated mitogenesis of endothelial cells
PDGFR. Inhibition of these two tyrosine kinases was
much more potently inhibited relative to either
found to be competitive with ATP, but the inhibition
PDGFR or FGFR) (Laird et al., 2000). Like
Tyrosine kinase inhibitors in cancer treatment trials
SU 5416, SU 6668 was found to be potent and
halted and marked regressions could again be induced
ecacious in a variety of tumor models. Unlike
in these tumors upon re-treatment. While the pre-
SU 5416, which was dosed i.p. in a DMSO-based
clinical data for both compounds appear to be very
vehicle, ecacy was achievable with SU 6668 when
promising, Phase I results for neither ZD4190 nor
dosed orally each day in a cremaphore-based vehicle.
In Phase I studies, SU 6668 was administered orally
once daily to 16 patients with advanced malignancies,
PTK 787 Novartis is reported to be developing PTK
at dose levels between 100 ± 1600 mg/m2/day (Rosen et
787, which has an anilinophthalazine pharmacophore
al., 2000). Nine of 16 patients remained on study for
(Bold et al., 2000) related to but distinct from the
up to 28 weeks while the remaining seven patients had
quinazolines described above (Figure 1). The com-
progressive disease. Dose limiting toxicities were not
pound is a potent inhibitor of both major human
observed, and dose escalation was said to be ongoing.
VEGFR (IC50 values of 37 and 77 nM for KDR and
Two patients at 1600 mg/m2 developed liver function
¯t-1, respectively) and, like STI 571, it provides potent
abnormalities, but both had potentially confounding
(sub-micromolar) inhibition of PDGFR and c-kit but
liver disease. Other possible drug related toxicities
does not inhibit v-abl, EGFR or FGFR (Wood et al.,
included nausea, headache, fatigue and changes in
2000). PTK 787 inhibits VEGF-induced KDR auto-
bowel movements. Pharmacokinetic data suggested
phosphorylation and mitogenesis, and promotes en-
that SU 6668 had a moderate-high clearance (78 l/
dothelial cell apoptosis, at a similar concentration
day/m2) and a somewhat improved elimination half-life
(Wood et al., 2000). The compound also has good
of 2.5 h relative to SU 5416 (Rosen et al., 2000). Phase
biopharmaceutical properties (plasma concentrations
II studies in multiple tumor types have apparently been
41 mM 8 h after administration of a 50 mg/kg oral
dose to mice), and impressive antiangiogenic
(ED50512.5 mg/kg/day for inhibition of angiogenesis
ZD4190 and ZD6474 ZD4190 is a quinazoline-based
in a s.c. growth factor implant model) and anti-tumor
VEGFR inhibitor (Figure 1) said to have entered
activity (signi®cant growth inhibition in six dierent
Phase I in early 2000. ZD6474 is thought to be from
human tumor xenograft models at daily oral doses of
the same structural class, but AstraZeneca has not yet
25 ± 75 mg/kg) (Wood et al., 2000). A key issue in the
disclosed the speci®c structure. ZD4190 inhibits both
®eld of anti-angiogenesis research has long been the
KDR and ¯t-1 (IC50 values of 29 and 708 nM,
fear that inhibition of tumor angiogenesis would also
respectively), and much less potent at inhibiting FGFR
impair normal angiogenesis, such as that in wound
(approximately 200-fold relative to KDR). The
healing. Given that most solid tumors are managed
compound is also 30-fold more potent at inhibiting
using multi-modality treatments that include surgery,
VEGF-mediated endothelial cell growth relative to
this has been a theoretical limitation to inhibitors of
FGF-stimulated cell growth (IC50 values of 50 and
angiogenesis. Interestingly, PTK 787 appears to have
1530 nM, respectively) (Wedge et al., 2000a). In vivo,
much less ecacy as an inhibitor of physiological
the compound was found to inhibit capillary invasion
angiogenesis of wound healing than as an eective
of cartilage (increased epiphyseal growth plate area),
blocker of tumor angiogenesis. Daily dosing of rats up
and to inhibit the growth of four human tumor
to 50 mg/kg day did not impair the healing or decrease
xenografts in a dose-dependent manner with daily oral
the tensile strength of full-thickness incisional wounds
administration (Wedge et al., 2000a). Direct measure-
(Wood et al., 2000). Data on the antitumor activity of
ments of tumor vascular endothelial permeability,
PTK 787 were recently extended to a renal tumor
using contrast medium-enhanced MRI indicated that
implant model, which was used to show that the
acute ZD4190 treatment produced measurable changes
compound could also inhibit both primary tumor
in vascular permeability at doses which yielded anti-
growth and the emergence of tumor metastasis to the
tumor activity during chronic administration (Wedge et
lung. Using a non-invasive (color Doppler imaging)
al., 1999). ZD6474, the second putative development
surrogate endpoint, a commensurate decrease in renal
candidate, is unique among small molecule angiogen-
artery blood ¯ow could also be observed after chronic
esis inhibitors, in that it is found to induce signi®cant
treatment (Drevs et al., 2000). Thus, PTK 787 appears
regressions in PC-3 tumors of varying size, with
to show signi®cant preclinical ecacy, and to produce
greatest eects being produced in the largest tumors
potent anti-tumor eects under well-tolerated dosing
(Wedge et al., 2000b). An intermittent ZD6474
regimens. The preclinical toxicological pro®le and the
treatment schedule, involving withdrawal of compound
human pharmacokinetics of this compound have not
for 4 weeks, revealed that tumor re-growth could be
Table 1 Selected small molecule tyrosine kinase inhibitors in clinical development for cancer
Phase II/III in multiple tumors (discontinued?)
Tyrosine kinase inhibitors in cancer treatment trials
directed. This may perhaps be a non-issue for VEGFR
and PDGFR inhibitors, which target transiently,
It is clear that the development of newer agents like the
focally activated receptors on normal cells. However,
tyrosine kinase inhibitors will require new concepts and
it is clear that there may be dierent approaches in
clinical paradigms that are distinctly dierent from
dealing with this issue during the development of
those used to develop the well-known cytotoxic agents
EGFR inhibitors. AstraZeneca has apparently not
commonly used in cancer chemotherapy. Some recent
incorporated prospective measurements of EGFR
commentaries have done an outstanding job at framing
over-expression in tumors from patients with NSCLC
these development issues (Sausville, 2000; Workman,
as an inclusion criteria in their Phase III trials with
2000; Hudes, 1999; Eisenhauer, 1998).
Iressa2. As mentioned previously, one can surmise that
Paramount among these is the need for non-
the rationale for this strategy was based on both the
conventional endpoints in clinical trial design, and for
high proportion of NSCLC tumors that over-express
the identi®cation, validation and implementation of
EGFR, and by preclinical data showing that over-
surrogate endpoints which may help direct dose-
expression is not predictive of a drug combination anti-
modulation during therapy. From the examples
provided above, it is clear that for several agents
P®zer and Oncogene Science have executed at least
(Iressa, CP-358,774; SU 5416), the presumed ecacious
one Phase II study with CP-358,774 in head and neck
dose in humans is very close to the maximally tolerated
cancer patients where EGFR expression levels were
dose. None of these agents has yet been in a clinical
evaluated as an entry criterion (Siu et al., 2000). The
trial designed to probe a broader aspect of ecacious
basis of this strategy could be said to reside in the
dose range. Given the somewhat poor performance of
Genentech development experience with Herceptin2, in
preclinical ecacy models in predicting eective plasma
which all patients entered into clinical trial, and
concentrations in humans, organizations developing
subsequently all patients receiving the approved
these new agents often resort to targeting some
commercial product, are ®rst pre-screened to detect
multiple of the plasma concentration required to
the level of over-expression of erbB2 in their breast
generate ecacy in animal models, without ®rst gaining
tumors. It is interesting to note that retrospective
an understanding as to whether clinical ecacy is dose-
analyses attempting to relate the level of erbB2
responsive, or that most patients are not being dosed at
expression to clinical response in patients treated with
a level well-along on the plateau of the dose-response
Herceptin2 (Dowsett et al., 2000) have been incon-
curve. Non-invasive approaches (Doppler and contrast
sistent and unconvincing. It is somewhat troubling to
agent imaging for VEGFR inhibitors) and invasive
see that so straight-forward an assay (immunohisto-
approaches (tumor and tissue sampling pre- and post-
chemistry) applied on a post hoc basis to understand an
treatment for EGFR inhibitors) are being developed to
agent with so singular a mechanism of action
aid in the assessment of minimally and maximally
(Herceptin2) has led to so little insight.
eective doses during the ®rst days and weeks of
One can perhaps begin to guage the obstacles that
clinical trial. The development of these surrogate
may lie ahead for the development of surrogate
endpoints is occurring on a parallel path with the
endpoints, which encompasses the application of novel
agents themselves, probably too late to help de®ne the
technologies applied in a prospective way to drugs with
dose-response, the minimally- and maximally-eective
complex mechanisms of action and pharmacological
dose, or the most ecient development paradigm.
eects. This is the next frontier for the development of
A second key issue is one of the biochemical
these new therapies for cancer. As such, the next 5 ± 6
selectivity of tyrosine kinase inhibitors, and the impact
years are likely to be as challenging, and as
that it may have on both the ecacy and the safety of
exhilarating, as have been the past 5 ± 6 years.
the clinical candidate. The current experience with both
non-selective tyrosine kinase inhibitors (STI 571 and
SU 5416) and selective compounds (Iressa2 and CP-
358,774) suggest that ecacy can be generated with
ATP, adenosine triphosphate; EGF/EGFR, epidermal
either class of inhibitor, with modest, comparable safety
growth factor/EGF receptor; PDGF/PDGFR, platelet-
margins. There could be an opportunity to assess the
derived growth factor/PDGF receptor; CML, chronic
relative merits of EGFR-selective vs. pan erbB
inhibitors when comparing the results of the trials with
Iressa2, CP-358,774 and CI-1033, but the irreversibility
of the latter candidate is likely to confound the
I would like to acknowledge the outstanding contributions
comparisons of relative therapeutic index. A related
of each member of the P®zer Global R&D division and the
issue is one of pre-screening patients for the over-
P®zer Oncology team for their insights, support, commit-
expression of the target at which the tyrosine kinase is
Allen LF, Cerna C, Gomez L, Yochmowitz M, Medina ML
Bartlett RR and Schleyerbach R. (1985). Intl. J. Immuno-
and Weitman S. (2000). Proc. NCI-EORTC-AACR
Symposium on New Drugs in Cancer Therapy, 384.
Baselga J, Herbst R, LoRusso P, Rischin D, Ranson M,
Ashman LK. (1999). Intl. J. Biochem. Cell Biol., 31, 1037 ±
Plummer R, Raymond E, Maddox A-M, Kaye SB,
Kieback DG, Harris A and Ochs J. (2000). Proc. Am.
Barbacci EG, Cunningham A, Iwata K, Moyer JD and
Miller PE. (1997). Proc. Am. Assoc. Cancer Res., 38, 3143.
Tyrosine kinase inhibitors in cancer treatment trials
Bold G, Altmann KH, Frei J, Lang M, Manley PW, Traxler
Heinrich MC, Grith DJ, Druker BJ, Wait CL, Ott KA and
P, Wietfeld B, Bruggen J, Buchdunger E, Cozens R,
Zigler AJ. (2000). Blood, 96, 925 ± 932.
Ferrari S, Furet P, Hofmann F, Martiny-Baron G, Mestan
Hudes G. (1999). J. Clin. Oncol., 17, 1093 ± 1094.
J, Rosel J, Sills M, Stover D, Acemoglu F, Boss E,
Hunter T. (2000). Cell, 100, 113 ± 127.
Emmenegger R, Lasser L, Masso E, Roth R, Schlachter C
Iwata K, Miller PE, Barbacci EG, Arnold LD, Doty J,
and Vetterli W. (2000). J. Med. Chem., 43, 2310 ± 2323.
DiOrio CI, Pustilnik LR, Reynolds M, Thelemann A,
Bonomi P, Perez-Soler R, Chachoua A, Huberman M, Karp
Sloan D and Moyer JD. (1997). Proc. Am. Assoc. Cancer
D, Rigas J, Hammond L, Rowinsky E, Preston G,
Ferrante KJ and Allen LF. (2000). Proc. NCI-EORTC-
Karp DD, Silberman SL, Csudae R, Wirth F, Gaynes L,
AACR Symposium on New Drugs in Cancer Therapy, 386.
Posner M, Bubley G, Koon H, Bergman M, Huang M and
Bridges AJ. (1999). Curr. Med. Chem., 6, 825 ± 843.
Schnipper LE. (1999). Proc. Am. Soc. Clin. Oncol., 18,
Cherrington JM, Strawn LM and Shawver LK. (2000). Adv.
Kelliher MA, McLaughlin J, Witte ON and Rosenberg N.
Ciardello F, Caputo R, Bianco R, Damiano V, Pomatico G,
(1990). Proc. Natl. Acad. Sci. USA, 87, 6649 ± 6653.
De Placido S, Bianco AR and Tortora G. (2000). Clin.
Kelly HC, Ferry D, Hammond L, Kris M, Ranson M and
Rowinsky E. (2000). Proc. Am. Assoc. Cancer Res., 41,
Cohen RB, Falcey JW, Paulter VJ, Fetzer KM and Waksal
HW. (2000a). Proc. Am. Soc. Clin. Oncol., 19, 1862.
Kuo EA, Hambleton PT, Kay DP, Evans PL, Matharu SS,
Cohen S, Smolen J, Emery E, Cannon G, Weaver A and
Little E, McDowall N, Jones CB, Hedgecock CJ, YeaCM,
Schi M. (2000b). Arthr. Rheum., 43 (Suppl. 9), 1221.
Chan AW, Hairsine PW, Ager IR, Tully WR, Wwilliason
Connell RD. (2000). Exp. Opin. Ther. Patents 10, 767 ± 786.
RA and Westwood R. (1996). J. Med. Chem., 39, 4608 ±
Cropp G, Rosen L, Mulay M, Langecker P and Hannah A.
(1999). Proc. Am. Soc. Clin. Oncol., 18, 619.
Kurzrock R, Gutterman JU and Talpaz M. (1988). N. Engl.
Dowsett M, Cooke T, Ellis I, Gullick WJ, Gusterson B,
Mallon E and Walker R. (2000). Eur. J. Cancer, 36, 170 ±
Laird AD, Vajkoczy P. Shawver LK, Thurnher A, Liang C,
Mohammadi M, Schlessinger J, Ullrich A, Hubbard SR,
Drevs J, Hofmann I, Hugenschmidt H, Wittig C, Madjar H,
Blake RA, Fong TAT, Strawn LM, Sun L, Tang C,
Muller M, Wood J, Martiny-Baron G, Unger C and
Hawtin R, Tang F, Shenoy N, Hirth KP, McMahon G and
Marme D. (2000). Cancer Res., 60, 4819 ± 4824.
Cherrington JL. (2000). Cancer Res., 60, 4152 ± 4160.
Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM,
Lawrence DS and Niu J. (1998). Pharmacol. Ther., 77, 81 ±
Fanning S, Zimmermann J and Lydon NB. (1996). Nat.
le Coutre P, Mologni L, Cleris L, Marchesi E, Buchdunger E,
Druker BJ and Lydon NB. (2000). J. Clin. Invest., 105, 3 ± 7.
Giardini R, Formelli F and Gambacorti-Passerini C.
Eckhardt SG, Rizzo J, Sweeney KR, Cropp G, Baker SD,
(1999). J. Natl. Cancer Inst., 91, 163 ± 168.
Kraynak MA, Kuhn JG, Villalona-Calero M, Hammond
Levitzki A and Bohmer FD. (1998). Anti-Cancer Drug
L, Weiss G, Thurman A, Smith L, Drengler R, Eckhardt
JR, Moczygemba J, Hannah AL, von Ho DD and
Levitzki A. (1999). Pharmacol. Ther., 82, 231 ± 239.
Rowinsky EK. (1999). J. Clin. Oncol., 17, 1095 ± 1104.
Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS,
Eisenhauer EA. (1998). Annals. Oncol., 9, 1047 ± 1052.
Jenkins NA. And Lee DC. (1994). Genes Dev., 8, 399 ± 413.
Ferry D, Hammond L, Ranson M, Kris MG, Miller V,
Mattar T, Kochhar K, Bartlett R, Bremer EG and Finnegan
Murray P, Tullo A, Feyereislova A, Averbuch S and
A. (1993). FEBS Lett., 334, 161 ± 164.
Rowinsky E. (2000). Proc. Am. Soc. Clin. Oncol., 19, 5E.
Medina L, Gomez L, Cerna C, Kraker A, Yochmowitz M
Fong TAT, Shawver LK, Sun L, Tang C, App H, Powell TJ,
and S Weitman S. (2000). Proc. Am. Assoc. Cancer Res.,
Kim YH, Schreck R, Wang X, Risau W, Ullrich A, Hirth
KP and McMahon G. (1999). Cancer Res., 59, 99 ± 106.
Mendel DB, Laird BD, Smolich BD, Blake RA, Liang C,
Fry DW. (2000). Anti-Cancer Drug Design, 15, 3 ± 16.
Hannah AL, Shaheen RM, Ellis LM, Weitman S, Shawver
Fry DW, Bridges AJ, Denny WA, Doherty A, Greis K, Hicks
LK and Cherrington JM. (2000). Anti-Cancer Drug
JL, Hook KE, Keller PR, Leopold WR, Loo J, Mcnamara
DJ, Nelson JM, Sherwood V, Smaill JB, Trumpp-
Moyer JD, Barbacci EG, Iwata KK, Arnold L, Boman B,
Kallmeyer S and Dobrusin E. (1998). Proc. Natl. Acad.
Cunningham A, DiOrio C, Doty J, Morin MJ, Moyer MP,
Neveu M, Pollack VA, Pustilnik LR, Reynolds MM,
Fry DW, Kraker AJ, McMichael A, Ambroso LA, Nelson
Sloan D, Theleman A and Miller P. (1997). Cancer Res.,
JM and Leopold WR. (1994). Science, 265, 1093 ± 1095.
Garber K. (2000). J. Natl. Cancer Inst., 92, 967 ± 969.
Nelson JM and Fry DW. (2000). Proc. Am. Assoc. Cancer
Ghosh S, Narla RK, Zheng Y, Liu X-P, Jun X, Mao C,
Sudbeck EA and Uckun FM. (1999). Anti-Cancer Drug
Osherov N and Levitzki A. (1994). Eur. J. Biochem., 225,
Gibbs JB. (2000). J. Clin. Invest., 105, 9 ± 13.
Plate KH, Breier G, Weich HA, Mennel HD and Risau W.
Goss G, Hirte H, Batist G, Stewart D, Miller W, Lorimer I,
(1994). Int. J. Cancer, 59, 520 ± 529.
Abugaber A, Matthews S and Seymour L. (2000). Proc.
Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan
DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA,
Hamby JM and Showalter HDH. (1999). Pharmacol. Ther.,
Davis JA, Vaidya MP, Arnold LD, Doty JL, Iwata K and
Morin MJ. (1999). J. Pharmacol. Exp. Ther., 291, 739 ±
Hamilton LC, Vojnovic I and Warner TD. (1999). Br. J.
Randal J. (2000). J. Natl. Cancer Inst., 92, 520 ± 522.
Hammond LA, Denis LJ, Salman UA, Chintapalli K,
Rosen L, Hannah A, Rosen P, Kabbinavar F, Mulay M,
Hidalgo M, Jeraback P, Patnaik A, Allen LF, Ferrante
Gicanov N, DePaoli A, Cropp G and Mabry M. (2000).
KJ, Carter WO, Kuhn, Drengler JR, Silberman S and
Proc. Am. Soc. Clin. Oncol., 19, 708.
Rowinsky EK. (2000). Proc. NCI-EORTC-AACR Sympo-
Rosen L, Mulay M, Mayers A, Kabbinavar F, Rosen P,
sium on New Drugs in Cancer Therapy, 385.
Cropp G and Hannah A. (1999). Proc. Am. Soc. Clin.
Hanahan D and Weinberg RA. (2000). Cell, 100, 57 ± 70.
Tyrosine kinase inhibitors in cancer treatment trials
Sausville EA. (2000). Anti-Cancer Drug Design, 15, 1 ± 2.
Wakeling AE, Barker AJ, Daview DH, Brown DS, Green
Sedlacek HH. (2000). Drugs, 59, 435 ± 476.
LR, Cartlidge SA and Woodburn JR. (1996). Breast
Shawver LK, Schwartz DP, Mann E, Chen H, Tsai J, Chu L,
Taylorson L, Longhi M, Meredith S, Germain L, Jacobs
Wang W-L, Healy ME, Sattler M, Verma S., Lin J, Maulik
JS, Tang C, Ullrich A, Berens ME, Hersh E, McMahon G,
G, Stiles CD, Grin JD, Johnson BE and Salgia R. (2000).
Hirth KP and Powell TJ. (1997). Clin. Cancer Res., 3,
Ward WHJ, Cook PN, Slater AM, Daview H, Holdgate GA
Sherwood V, Bridges AJ, Denny WA, Rewcastle GW, Smaill
and Green LR. (1994). Biochem. Pharmacol., 48, 659 ± 666.
JB and Fry DW. (1999). Proc. Am. Assoc. Cancer Res., 40,
Wedge SR, Waterton JC, Tessier JJ, Checkley D, Dukes M,
Kendrew J and Curry B. (1999). Proc. Am. Assoc. Cancer
Sirotnak FM, Zakowsky MF, Miller VA, Scher HI and Kris
MG. (2000). Proc. Am. Assoc. Cancer Res., 41, 3076.
Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Curwen, JO,
Siu LL, Hidalgo M, Nemunaitis J, Rizzo J, Moczygemba J,
Hennequin F, Thomas AP, Stokes ESE, Curry B,
Eckhardt SG, Tolcher A, Smith L, Hammond L, Black-
Richmond GHP and Wadsworth PF. (2000a). Cancer
burn A, Tensfeldt T, Silberman S and von Ho DD.
(1999). Proc. Am. Soc. Clin. Oncol., 18, 1498.
Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Hennequin F,
Siu LL, Soulieres D, Senzer N, Agarwala S, Vokes E, Fisher
Stokes ESE and Curry B. (2000b). Proc. Am. Assoc.
D, Marsolais C, Ferrante KJ and Allen LF. (2000). Proc.
NCI-EORTC-AACR Symposium on New Drugs in Cancer
Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei
J, Hofmann F, Mestan J, Mett H, O'Reilly T, Persohn E,
Smolen JS and Emery P. (2000). Rheumatol., 39 (Suppl. 1),
Rosel J, Schnell C, Stover D, Theuer A, Towbin H,
Wenger F, Woods-Cook K, Menrad A, Siemeister G,
Songyang Z and Cantley LC. (1998). Methods Mol. Biol., 87,
Schirner M, Thierauch K-H, Schneider MR, Drevs J,
Martiny-Baron G, Totzke F and Marme D. (2000). Cancer
Sungaran R, Chislom OT, Markovic B, Khachigian LM,
Tanaka Y and Chong BH. (2000). Blood, 95, 3094 ± 3101.
Woodburn JR, Barker AJ, Gibson KH, Ashton SE,
Talpaz M, Sawyers CL, Kantarjain H, Resta D, Fernandes
Wakeling AE, Curry BJ, Scarlett L and Henthord LR.
Reese S, Ford J and Druker BJ. (2000). Proc. Am. Soc.
(1997). Proc. Am. Assoc. Cancer Res., 38, 4251.
Workman P. (2000). Curr. Opin. Oncol., Endocrin. Metab.
Thompson WD, Li WW and Maragoudakis M. (1999). J.
Xu X, Williams JW, Bremer EG, Finnegan A and Chong AS-
Tzahar E, Waterman H, Chen XM, Levkowitz G, Karuna-
F. (1995). J. Biol. Chem., 270, 12398 ± 12403.
garan D, Lavi S, Ratzkin BJ and Yarden Y. (1996). Mol.
Vincent PW, Patmore SJ, Atkinson BE, Bridges AJ, Kirkish
LS, Dudeck RC, Leopold WR, Zhou H and Elliott WL.
(1999). Proc. Am. Assoc. Cancer Res., 40, 117.
Devon & Cornwall Police Record 1 Freedom of Information Act Request No: 007722/11 1. A copy of the renewal reminder letters sent to holders of firearm and shot gun certificates. If such reminders are not routinely sent please explain why the ACPO guidance is not followed. 2. The time in weeks prior to the renewal date at which the reminder letter is The
Optimal Competitive Online Ray Search with an Error-Prone Robot University of Bonn, Institute of Computer Science I { kamphans,langetep } @cs.uni-bonn.de Abstract. We consider the problem of finding a door along a wall with a blind robot that neither knows the distance to the door nor the direc- tion towards of the door. This problem can be solved with the well- known doubling strategy yi
 Tyrosine kinase inhibitors in cancer treatment trials
Figure 1 Structures of selected tyrosine kinase inhibitors in clinical development for cancer
nonetheless, progressed into clinical trials by SUGEN
clinical ®ndings with AstraZeneca's ZD1839 (Iressa2)
(now part of Pharmacia). A Phase I study in cancer
have been equally compelling. The pharmacological
patients revealed that SU 101 was well-tolerated as a
characteristics of Iressa2 were ®rst described in 1996
24 h continuous i.v. infusion at doses up to 443 mg/m2/
(Wakeling et al., 1996; Woodburn et al., 1997) as a
wk. At this dose, the plasma concentration of the
potent and selective inhibitor of the EGFR tyrosine
active metabolite was maintained at levels sucient to
kinase. This quinazoline-based compound (Figure 1) is
block both PDGFR and EGFR signaling, as well as
an ATP-competitive inhibitor of the EGFR tyrosine
pyrimidine biosynthesis (Eckhardt et al., 1999). Toxi-
kinase (IC50 25 nM) with 50-fold selectivity relative to
cities were relatively minor (Grade 1 ± 2 nausea,
closely homologous erbB family members (IC50 for
vomiting and fever in approximately 20% of all
erbB2 1 ± 3 mM) and even greater selectivity for more
courses given). Surprisingly, hematopoietic toxicities
divergent tyrosine kinases. It demonstrates good
and hemolysis, which had been noted in the preclinical
cellular potency (80 nM IC50 for inhibition of EGF-
experience with SU 101, were not seen in this Phase I
dependent mitogenesis) and robust, dose-dependent
population. One partial response was seen in 26
anti-tumor ecacy in a variety of human tumor
patients receiving an average of two courses each; the
xenografts (Woodburn et al., 1997). These results have
responding patient received 13 courses (52 infusions) to
been most recently extended to show that Iressa2 has
treat an anaplastic astrocytoma, and had a notable
in vivo ecacy in a diverse human tumor xenograft
(450%) reduction in one measurable lesion (Eckhardt
models both with (Ciardello et al., 2000) and without
et al., 1999). SU 101 has been reported to be in
(Sirotnak et al., 2000) highly activated EGFR signaling
advanced trials for multiple solid tumor types, but
pathways. Of equal interest are the observations that
recent disclosures (Garber, 2000) indicate that Phase
Iressa2 combines with standard cytotoxic agents
III trials in at least one tumor type (glioblastoma) have
(platinums, taxanes, topoisomerase I inhibitors, etc.)
been abandoned. The status of other trials (ongoing
to produce additive or supra-additive anti-tumor
Phase II trials for ovarian and NSCLC; planned Phase
ecacy in vivo without exacerbation of the toxicity of
III trials for prostate, colon and NSCLC) is uncertain
the co-administered cytotoxics. The ®ndings that tumor
EGFR density does not predict ecacy when the
compound is used in conjunction with cytotoxic agents
have signi®cantly impacted the development strategy
employed by AstraZeneca as Iressa2 moves towards
Iressa2 (ZD1839) While STI 571 has provided no-
Multiple Phase I trials with Iressa2 have been
table clinical proof-of-concept for the clinical ecacy
summarized, and the results revealed reasonable
and safety of tyrosine kinase inhibitors, the early
pharmacokinetics, good toleration and the ®rst signs
Tyrosine kinase inhibitors in cancer treatment trials
of clinical ecacy when used as a single agent in
(Pollack et al., 1999; Goss et al., 2000; Allen et al.,
patients with advanced disease (Ferry et al., 2000;
2000). Given the overall safety and toleration pro®le of
Baselga et al., 2000; Kelly et al., 2000). Following oral
Iressa2, AstraZeneca has committed to an aggressive
administration of a single dose (50 mg), maximum
development strategy, which includes two large Phase
plasma drug concentrations (mean 45 ng/ml) occurred
III studies to assess the use of Iressa2 in combination
1 ± 5 h post-dose. The mean terminal t1/2 was 34 h.
Tyrosine kinase inhibitors in cancer treatment trials
Figure 1 Structures of selected tyrosine kinase inhibitors in clinical development for cancer
nonetheless, progressed into clinical trials by SUGEN
clinical ®ndings with AstraZeneca's ZD1839 (Iressa2)
(now part of Pharmacia). A Phase I study in cancer
have been equally compelling. The pharmacological
patients revealed that SU 101 was well-tolerated as a
characteristics of Iressa2 were ®rst described in 1996
24 h continuous i.v. infusion at doses up to 443 mg/m2/
(Wakeling et al., 1996; Woodburn et al., 1997) as a
wk. At this dose, the plasma concentration of the
potent and selective inhibitor of the EGFR tyrosine
active metabolite was maintained at levels sucient to
kinase. This quinazoline-based compound (Figure 1) is
block both PDGFR and EGFR signaling, as well as
an ATP-competitive inhibitor of the EGFR tyrosine
pyrimidine biosynthesis (Eckhardt et al., 1999). Toxi-
kinase (IC50 25 nM) with 50-fold selectivity relative to
cities were relatively minor (Grade 1 ± 2 nausea,
closely homologous erbB family members (IC50 for
vomiting and fever in approximately 20% of all
erbB2 1 ± 3 mM) and even greater selectivity for more
courses given). Surprisingly, hematopoietic toxicities
divergent tyrosine kinases. It demonstrates good
and hemolysis, which had been noted in the preclinical
cellular potency (80 nM IC50 for inhibition of EGF-
experience with SU 101, were not seen in this Phase I
dependent mitogenesis) and robust, dose-dependent
population. One partial response was seen in 26
anti-tumor ecacy in a variety of human tumor
patients receiving an average of two courses each; the
xenografts (Woodburn et al., 1997). These results have
responding patient received 13 courses (52 infusions) to
been most recently extended to show that Iressa2 has
treat an anaplastic astrocytoma, and had a notable
in vivo ecacy in a diverse human tumor xenograft
(450%) reduction in one measurable lesion (Eckhardt
models both with (Ciardello et al., 2000) and without
et al., 1999). SU 101 has been reported to be in
(Sirotnak et al., 2000) highly activated EGFR signaling
advanced trials for multiple solid tumor types, but
pathways. Of equal interest are the observations that
recent disclosures (Garber, 2000) indicate that Phase
Iressa2 combines with standard cytotoxic agents
III trials in at least one tumor type (glioblastoma) have
(platinums, taxanes, topoisomerase I inhibitors, etc.)
been abandoned. The status of other trials (ongoing
to produce additive or supra-additive anti-tumor
Phase II trials for ovarian and NSCLC; planned Phase
ecacy in vivo without exacerbation of the toxicity of
III trials for prostate, colon and NSCLC) is uncertain
the co-administered cytotoxics. The ®ndings that tumor
EGFR density does not predict ecacy when the
compound is used in conjunction with cytotoxic agents
have signi®cantly impacted the development strategy
employed by AstraZeneca as Iressa2 moves towards
Iressa2 (ZD1839) While STI 571 has provided no-
Multiple Phase I trials with Iressa2 have been
table clinical proof-of-concept for the clinical ecacy
summarized, and the results revealed reasonable
and safety of tyrosine kinase inhibitors, the early
pharmacokinetics, good toleration and the ®rst signs
Tyrosine kinase inhibitors in cancer treatment trials
of clinical ecacy when used as a single agent in
(Pollack et al., 1999; Goss et al., 2000; Allen et al.,
patients with advanced disease (Ferry et al., 2000;
2000). Given the overall safety and toleration pro®le of
Baselga et al., 2000; Kelly et al., 2000). Following oral
Iressa2, AstraZeneca has committed to an aggressive
administration of a single dose (50 mg), maximum
development strategy, which includes two large Phase
plasma drug concentrations (mean 45 ng/ml) occurred
III studies to assess the use of Iressa2 in combination
1 ± 5 h post-dose. The mean terminal t1/2 was 34 h.